Presentación de Caso
Un hombre de 55 años fue trasladado a este hospital
para la evaluación de una masa mediastínica que se identificó como parte de una
evaluación de fiebres intermitentes, sudores nocturnos y fatiga.
El paciente había estado bien hasta 3 meses antes de
la admisión a este hospital, cuando desarrolló fatiga y una erupción
maculopapular pruriginosa transitoria en el antebrazo. Durante las siguientes 6
semanas, la erupción del antebrazo se resolvió pero la fatiga persistió. El
paciente comenzó a dormir siestas durante el día y no podía dedicar días
completos a su trabajo en la construcción. También comenzó a tener sudores
nocturnos y fiebres con temperaturas de hasta 37,8°C.
Veintisiete días antes de la admisión a este hospital,
el paciente descubrió que no podía trabajar debido a la fatiga y buscó una
evaluación en el departamento de emergencias de otro hospital. Los análisis de
sangre para ácidos nucleicos de anaplasma y babesia fueron negativos, pero las
pruebas de detección y confirmación para la enfermedad de Lyme IgM e IgG fueron
positivas. Se realizó el diagnóstico de enfermedad de Lyme y se inició
tratamiento con doxiciclina. Debido a que se desarrolló malestar
gastrointestinal, se suspendió la doxiciclina y se prescribió amoxicilina por
el resto del curso de 14 días.
Dieciséis días antes de la admisión a este hospital,
el paciente comenzó a tener molestias en el pecho, que describió como una
sensación de “pelota de softball” en la parte inferior izquierda del pecho. Las
molestias en el pecho limitaban su capacidad para respirar y su médico de
atención primaria lo derivó al departamento de emergencias del otro hospital.
En la evaluación refirió fatiga y malestar creciente, sudoración nocturna y
fiebre, junto con artralgias y mialgias difusas, cefalea, fotofobia con algunos
destellos visuales, disnea de esfuerzo, edema, erupción eritematosa pruriginosa
en el tórax, anorexia, náuseas, vómitos , malestar y distensión abdominal, y
diarrea acuosa que se presentaba tres veces al día.
En el examen, la temperatura era de 36,8°C, la presión
arterial de 145/84 mm Hg, la frecuencia cardíaca de 61 latidos por minuto y la
saturación de oxígeno del 99% mientras el paciente respiraba aire ambiente. El
peso fue de 83,9 kg y el índice de masa corporal de 28,1. Había un exantema
macular eritematoso difuso tenue en la porción anterior del tórax. El abdomen
estaba levemente distendido y difuso a la palpación. Los niveles sanguíneos de
alanina aminotransferasa, aspartato aminotransferasa, bilirrubina y lipasa
fueron normales; otros resultados de pruebas de laboratorio se muestran en la
Tabla 1. Se obtuvo sangre para cultivo microbiológico. Un electrocardiograma
fue normal. Se administró suero salino intravenoso y paracetamol oral, así como
tratamiento empírico con piperacilina-tazobactam intravenoso, hidromorfona y ondansetrón.
El paciente fue ingresado en el otro hospital.


Tabla 1. Datos de laboratorio.
Quince días antes de la admisión a este hospital, una
tomografía computarizada (TC) de tórax, abdomen y pelvis ( Figura 1A, 1B y 1C
), realizada sin la administración de material de contraste intravenoso, reveló
una masa heterogénea de partes blandas, que medía 6,1 cm por 2,8 cm, en el
mediastino anterior. Había múltiples ganglios linfáticos subcarinales y
mediastínicos aumentados de tamaño (que medían hasta 15 mm en el eje corto de
diámetro), múltiples ganglios linfáticos mesentéricos aumentados de tamaño (que
medían hasta 11 mm en el eje corto de diámetro), infiltración de la grasa
mesentérica y trazas de ascitis.

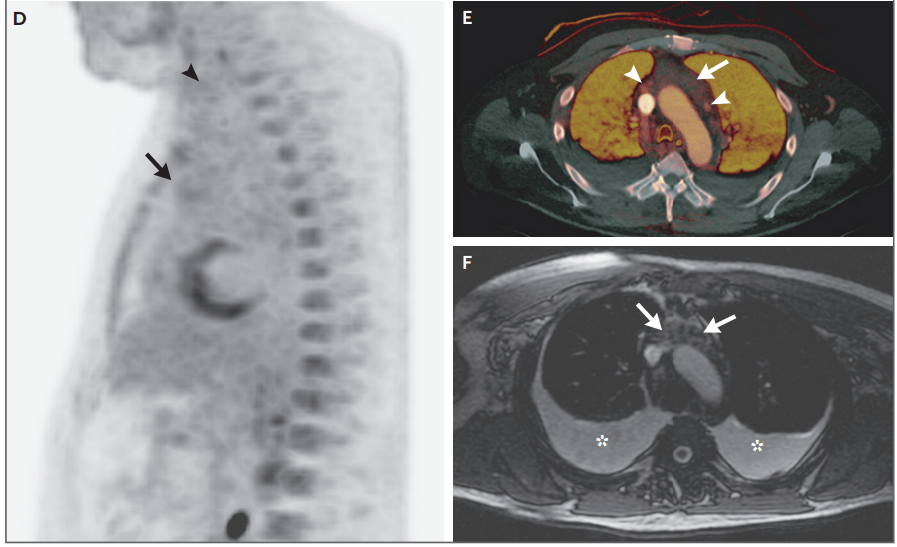
Figura 1. Estudios de imagen.
Quince días antes del ingreso en este hospital, una
TAC de tórax, abdomen y pelvis se realizó en el primer hospital. Una imagen
coronal (Panel A) muestra esplenomegalia (longitud del bazo, 14 cm; valor
normal, ≤12), sin infiltración grasa focal y trazas de ascitis (asteriscos) a
lo largo del mesenterio sigmoideo derecho y la fascia renal anterior izquierda.
Las imágenes transaxiales y coronales (Paneles B y C, respectivamente) muestran
una masa mediastínica anterior bilobulada, de 6,1 cm por 2,8 cm, con atenuación
de tejido blando (flechas) y ganglios linfáticos agrandados (puntas de flecha).
Nueve días antes de la admisión a este hospital, una tomografía por emisión de positrones con
18F-fluoro- desoxiglucosa (FDG-PET) se realizó en el segundo hospital. Un corte
grueso, proyección sagital de intensidad máxima de atenuación corregida en las
imágenes de PET (Panel D) muestran niveles
bajos de captación de FDG en la masa mediastínica anterior (flecha) y un
ganglio linfático cervical izquierdo agrandado (punta de flecha). El primer día
en este hospital se realizó una TC realzada con contraste de energía dual. Un
mapa de yodo transaxial (Panel E) muestra un realce mínimo de la parte anterior
de la masa mediastínica, que mide 6,1 cm por 1,9 cm (flecha), en comparación
con el realce prominentes de los ganglios
linfáticos mediastínicos (puntas de flecha). El día 13 en este hospital, se
comenzó a realizar la resonancia magnética, pero no se pudo completar debido a
que el paciente tenía dificultad respiratoria cuando fue colocado en decúbito
prono. Una imagen rápida transaxial empleando imagen de adquisición de estado
estacionario (FIESTA) (Panel F) muestra grasa interdigitada y tejido blando
hipointenso en comparación con el músculo, en el mediastino anterior (flechas).
La dificultad respiratoria fue causada por grandes derrames pleurales
bilaterales (asteriscos).
El malestar abdominal persistió; Se suspendió
piperacilina-tazobactam y se pautó ceftriaxona. Dos días después, la TC de
abdomen, realizada sin la administración de material de contraste intravenoso,
reveló un nuevo edema pulmonar, nuevos derrames pleurales bilaterales pequeños
y un aumento del volumen de la ascitis. El paciente fue trasladado a un segundo
hospital para una evaluación adicional.
En la exploración física destacaba leve dolor
abdominal y distensión, así como edema en las piernas. Los niveles sanguíneos
de alanina aminotransferasa, aspartato aminotransferasa y bilirrubina y un
análisis de orina fueron normales; en la Tabla 1 se muestran otros resultados
de pruebas de laboratorio . La tomografía por emisión de positrones (PET) con
18 F-fluorodesoxiglucosa (FDG) ( Figura 1D ) reveló una captación indeterminada
de FDG de bajo nivel en el tejido blando mediastínico anterior, así como en
derrames pleurales y ganglios linfáticos mediastínicos.
Durante la semana siguiente persistió la disnea y el
dolor abdominal. Aumento de edema, específicamente en abdomen, piernas y cara;
el peso aumentó a 93,0 kg. Se administraron ceftriaxona y metoclopramida por
vía intravenosa e hidromorfona, ondansetrón y lactulosa por vía oral. Se
realizaron pruebas de laboratorio adicionales ( Tabla 2 ).





Tabla 2. Datos de laboratorio adicionales.
El octavo día en el segundo hospital se realizó
cirugía torácica asistida por video. Según los informes, el mediastino apareció
inflamado. Al paciente se le realizó evacuación de líquido pleural y biopsia de
partes blandas mediastínicas y ganglios linfáticos principalmente en ventana
subcarinal (nivel VII). En el examen patológico de las muestras de biopsia, el
tejido blando mediastínico mostró tejido fibroconectivo y adiposo con fibrosis,
inflamación leve y necrosis grasa, proliferación capilar prominente y
congestión; Según los informes, los ganglios linfáticos eran "levemente
anormales". La evaluación microbiológica de las muestras de biopsia reveló
leucocitos ocasionales, pero no bacilos acidorresistentes ni elementos
fúngicos. En la citometría de flujo de las muestras de biopsia, el tejido
blando mediastínico no mostró ninguna población celular anormal; los ganglios
linfáticos contenían células B policlonales y poblaciones heterogéneas de
células T, sin evidencia inmunofenotípica de linfoma. Los cultivos de
bacterias, hongos y micobacterias de las muestras de biopsia fueron negativos.
El examen citológico del líquido pleural reveló células mesoteliales,
linfocitos e histiocitos reactivos, pero no células malignas.
Después del procedimiento, el nivel de creatinina
aumentó a 2,93 mg por decilitro (rango de referencia, 0,50 a 1,30). El examen
del sedimento urinario reveló cilindros hialinos y granulares. Se administró
furosemida intravenosa. El día 15 en el segundo hospital, el paciente fue
trasladado a este hospital.
A su llegada, el paciente refirió fatiga persistente,
disnea leve, distensión abdominal, edema y anorexia. Las náuseas, los vómitos,
la diarrea y el sarpullido en el pecho se habían resuelto. Las fiebres y los
sudores nocturnos se habían producido en un patrón creciente y menguante
durante las semanas anteriores. Refería fotofobia intermitente y cefalea, sin
rigidez de nuca ni fonofobia, y no presenta otros síntomas neurológicos.
El paciente no tenía antecedentes de enfermedad y no
había recibido atención médica de rutina como adulto. Tomaba ibuprofeno y
paracetamol según lo necesitaba y no tenía alergias conocidas a medicamentos.
Había estado expuesto al polvo a través de su trabajo en la construcción. Vivía
con su esposa, su hijo y su perro y gato en una comunidad boscosa costera en
Nueva Inglaterra. Con frecuencia pasaba tiempo al aire libre, ya sea trabajando
o jugando al golf, y había recibido múltiples picaduras de garrapatas. Había
viajado a las regiones del Mediterráneo y Oceanía en años anteriores y, más
recientemente, al suroeste de los Estados Unidos. El paciente nunca había
consumido tabaco ni drogas ilícitas y rara vez bebía cerveza. Su padre había
tenido cáncer de próstata y cáncer de colon.
En el examen, la temperatura era de 36,8°C, la presión
arterial de 148/82 mm Hg, la frecuencia cardíaca de 78 latidos por minuto, la
frecuencia respiratoria de 18 por minuto y la saturación de oxígeno del 93%
mientras el paciente respiraba aire ambiente. El peso era de 91,7 kg. El examen
neurológico era normal y no había rigidez de nuca. Había crepitantes
inspiratorios en el pulmón izquierdo. El abdomen estaba levemente distendido y
difuso a la palpación. Se notó una leve fluctuación en el lado derecho del
tórax, en el sitio de la incisión quirúrgica. No había adenopatías cervicales,
supraclaviculares, submandibulares, submentonianas, axilares o inguinales
palpables. Había 2+ edema con fóvea en las piernas.
Los niveles sanguíneos de alanina aminotransferasa,
aspartato aminotransferasa, bilirrubina, ceruloplasmina, fibrinógeno, folato,
globulina, haptoglobina y vitamina B 12 fueron normales; en la Tabla 1 se
muestran otros resultados de pruebas de laboratorio . El análisis de orina
mostró orina amarilla turbia con 2+ de proteína y de 3 a 5 glóbulos rojos por
campo de alta potencia (rango de referencia, 0 a 2). Un electrocardiograma fue
normal.
La TC de tórax, abdomen y pelvis ( Figura 1E ),
realizada después de la administración de material de contraste intravenoso,
reveló una masa en el mediastino anterior, que medía 6,1 cm por 1,9 cm, que era
ligeramente más pequeña de lo que había sido en el TAC de tórax previo. Había
agrandamiento de los ganglios linfáticos mediastínicos perivasculares, paratraqueales,
subcarinales y aortopulmonares (que medían hasta 12 mm en el eje corto del
diámetro) con realce heterogéneo. Se detectó aumento de edema pulmonar,
derrames pleurales bilaterales moderados, ascitis de volumen moderado,
esplenomegalia leve y múltiples ganglios mesentéricos subcentimétricos.
En el tercer día de hospitalización, la resonancia
magnética nuclear (RMN) de la cabeza no reveló anomalías. Al cuarto día, un
ecocardiograma transtorácico fue normal. Se administró furosemida intravenosa y
paracetamol oral. Se realizó punción lumbar y se extrajeron 26 ml de líquido
incoloro; no se midió la presión de apertura. Los resultados del análisis del
líquido cefalorraquídeo se muestran en la Tabla 3. Al quinto día, la ecografía
renal reveló riñones de tamaño normal con parénquima normal y sin
hidronefrosis. Se realizó paracentesis diagnóstica; los resultados del análisis
del líquido ascítico se muestran en la Tabla 3.

Tabla 3. Resultados del estudio del líquido ascítico y del LCR.
El día 13, se inició la resonancia magnética del
tórax, pero no se pudo completar porque el paciente tenía dificultad
respiratoria cuando se colocó en posición prona. Las secuencias que se
obtuvieron antes de la finalización del estudio ( Figura 1F ) mostraron una
masa de tejido blando en el mediastino anterior con hipointensidad en las
imágenes de eco de gradiente en estado estacionario; la masa era más pequeña de
lo que había sido en la TC de tórax realizada antes del traslado a este
hospital. Además, había ganglios linfáticos mediastínicos agrandados, grandes
derrames pleurales bilaterales, ascitis y un hematoma en el sitio de la
incisión quirúrgica.
Se realizaron pruebas de laboratorio adicionales (
Tabla 2 ). Se hizo un diagnóstico.
DIAGNÓSTICO DIFERENCIAL
Este hombre de 55 años previamente sano se presentó
con una masa mediastínica anterior, linfadenopatía mediastínica con baja
captación de FDG, ganglios linfáticos mesentéricos prominentes y esplenomegalia
en presencia de síntomas constitucionales, anasarca, niveles elevados de
inflamación marcadores, disfunción renal, hipoalbuminemia, anemia y
trombocitopenia. Su síndrome inflamatorio podría ser compatible con una
tormenta de citoquinas, que se caracteriza por niveles elevados de citoquinas
circulantes, síntomas inflamatorios sistémicos agudos y disfunción orgánica
debido a una respuesta inmune excesiva. 1Con una tormenta de citoquinas, la
identificación rápida de la causa subyacente y el inicio del tratamiento
apropiado son críticos. Los niveles elevados de marcadores inflamatorios,
incluida la tasa de sedimentación de eritrocitos y los niveles de proteína C
reactiva, ferritina e interleucina-6, son hallazgos inespecíficos. Sin embargo,
el desarrollo de este síndrome inflamatorio, junto con una masa mediastínica y
linfadenopatía, en un paciente inmunocompetente ayuda a reducir el diagnóstico
diferencial de cáncer, enfermedades infecciosas, enfermedades autoinmunes y
otras causas relacionadas con el sistema inmunitario.
CÁNCER
La constelación de una masa mediastínica,
linfadenopatía, esplenomegalia y síntomas constitucionales es más preocupante
para el linfoma, en particular el linfoma de Hodgkin, así como para el timoma y
otros tumores sólidos con predilección por el mediastino. Nos dijeron que los
ganglios linfáticos extirpados se habían caracterizado como "levemente
anormales" en el examen patológico, pero no recibimos una descripción
detallada de los hallazgos. Sin embargo, no se mencionó evidencia morfológica o
inmunofenotípica de células de Reed-Sternberg u otras poblaciones clonales.
Además, el examen patológico de la masa mediastínica no reveló evidencia
morfológica de timoma u otros tumores sólidos. La baja captación de FDG también
sugiere que el cáncer es poco probable en este caso. Sin embargo, los errores
de muestreo de biopsia pueden causar resultados falsos negativos,2 Dado que el
examen histológico de una muestra de tejido adecuada es la piedra angular del
diagnóstico de cáncer, necesitaríamos volver a examinar las muestras de biopsia
de este paciente para determinar si son suficientes o si es necesario repetir
la biopsia por escisión.
ENFERMEDADES INFECCIOSAS
Varias enfermedades infecciosas pueden causar un
síndrome inflamatorio sistémico, pero la presencia de linfadenopatía y una masa
mediastínica reduce la lista de posibilidades. Muchas enfermedades infecciosas,
incluidas infecciones bacterianas (p. ej., infección por Mycobacterium
tuberculosis u otras micobacterias, Borrelia burgdorferi u otros patógenos
transmitidos por garrapatas, Bartonella henselae , Treponema pallidum ,
Coxiella burnetiio leptospira), infecciones virales (p. ej., infección por el
virus de Epstein-Barr, citomegalovirus, virus de la inmunodeficiencia humana
[VIH] o virus de la hepatitis B) y otras infecciones (p. ej., toxoplasmosis,
histoplasmosis, criptococosis, coccidioidomicosis o paludismo) — son poco
probables, dadas las pruebas microbiológicas negativas, la presentación clínica
y la falta de respuesta al tratamiento antibiótico empírico. Aunque las pruebas
para la enfermedad de Lyme IgM e IgG fueron positivas, es muy poco probable que
la enfermedad de Lyme sea la causa subyacente del síndrome de este paciente,
dado que no tenía los rasgos característicos de la enfermedad, así como la
falta de respuesta a las semanas. de tratamiento antibiótico. Es importante
tener en cuenta que las pruebas para la enfermedad de Lyme IgM pueden
permanecer positivas durante años después de la infección. 3,4
ENFERMEDADES AUTOINMUNES
Las enfermedades autoinmunes, como el lupus
eritematoso sistémico, la artritis reumatoide, el síndrome de Sjögren, la
vasculitis, la miastenia grave, la enfermedad de Goodpasture y la fiebre
reumática, se pueden descartar por los síntomas de presentación y las pruebas
negativas de anticuerpos antinucleares y otros marcadores. Las mialgias y
artralgias del paciente probablemente reflejan una inflamación inespecífica.
Aunque el lupus se asocia con una variedad de síntomas y signos que se
superponen con las características de este caso, la prueba negativa de
anticuerpos antinucleares y la ausencia de hipocomplementemia proporcionan un
alto valor predictivo negativo, lo que nos permite descartar lupus en este
caso. 5,6
OTRAS CAUSAS RELACIONADAS CON LA INMUNIDAD
Otras enfermedades relacionadas con el sistema
inmunitario, como la sarcoidosis, la enfermedad relacionada con IgG4, la
amiloidosis y la deficiencia de la subclase de IgG2, son diagnósticos poco
probables en este caso, dados los hallazgos de imágenes, biopsia y laboratorio.
Las inmunodeficiencias hereditarias y las condiciones autoinflamatorias
normalmente no se manifestarían a los 55 años de edad. La sarcoidosis es una
consideración seria en este paciente, pero la ausencia de granulomas en el
tejido extirpado hace que este diagnóstico sea poco probable. Sin embargo, es
importante asegurarse de que las muestras de la biopsia sean suficientes para
descartar este diagnóstico, así como el cáncer. Aunque los niveles bajos de IgG
y la subclase IgG2 de este paciente podrían indicar una deficiencia de la
subclase IgG2, no tenía antecedentes de infección, y la ausencia de tales
antecedentes sugiere que estos hallazgos de laboratorio no son clínicamente
significativos. 7 Otras dos enfermedades relacionadas con el sistema
inmunitario permanecen en el diagnóstico diferencial.
LINFOHISTIOCITOSIS HEMOFAGOCÍTICA
La constelación de fiebres, esplenomegalia, citopenias
y niveles elevados de marcadores inflamatorios es característica de la
linfohistiocitosis hemofagocítica (HLH), que comprende un grupo de trastornos
raros de hiperactivación inmunitaria desencadenada por infección, cáncer o
enfermedades autoinmunes. Sin embargo, este paciente tenía un nivel de
fibrinógeno normal y un nivel de ferritina de menos de 1000 μg por litro, lo
que sería poco común en un paciente con HLH. 8 La linfadenopatía y una masa
mediastínica también serían inusuales en un paciente con HLH, a menos que la
HLH esté desencadenada por un linfoma o una infección, lo cual es poco probable
en este caso. No obstante, sería útil evaluar las características morfológicas
de la médula ósea, la actividad de las células asesinas naturales y los niveles
del receptor de interleucina-2 soluble. 9La evidencia de hemofagocitosis en la
médula ósea y la disminución de la actividad de las células asesinas naturales
respaldarían el diagnóstico de HLH; un nivel levemente elevado de receptor de
interleucina-2 soluble, que refleja la activación de células T, es muy sensible
pero menos específico para HLH. 10
ENFERMEDAD DE CASTLEMAN
La enfermedad de Castleman describe un grupo de
trastornos linfoproliferativos policlonales raros y heterogéneos que se asocian
con un espectro de rasgos histopatológicos característicos. Estas
características a menudo se clasifican como hipervasculares o vasculares
hialinas, plasmocíticas o mixtas. El síndrome de este paciente es muy
compatible con la enfermedad de Castleman multicéntrica (MCD), que implica
linfadenopatía multicéntrica, síntomas constitucionales, citopenias, anasarca y
disfunción orgánica debido a una tormenta de citocinas, a menudo impulsada por
la interleucina-6. La MCD puede resultar de una infección no controlada por el
herpesvirus humano 8 (HHV-8) en personas con infección por VIH o un estado
inmunocomprometido (MCD asociada a HHV-8), puede deberse a la producción de
citoquinas por células plasmáticas monoclonales (POEMS [polineuropatía,
organomegalia, endocrinopatía , proteína M y cambios en la piel], MCD
asociada), o puede tener una causa desconocida o incierta (MCD idiopática
[iMCD]). 11 Dado que se presume que este paciente es inmunocompetente y no hay
evidencia de células plasmáticas monoclonales, la MCD asociada a HHV-8 y la MCD
asociada a POEMS son poco probables; sin embargo, el tejido del ganglio linfático
extirpado debe analizarse para detectar HHV-8.
Este paciente cumple con todos los criterios de
diagnóstico de consenso para iMCD excepto por las características histológicas
en el tejido de los ganglios linfáticos, por lo que se justifica una segunda
revisión de la muestra. 12 Tiene linfadenopatía multicéntrica, así como los
siguientes 8 de 11 criterios menores (con diagnóstico que requiere ≥2): un
nivel elevado de proteína C reactiva, anemia, trombocitopenia, hipoalbuminemia,
disfunción renal, síntomas constitucionales, esplenomegalia y acumulación de
líquido. Además, se han descartado enfermedades que simulan iMCD. Aunque no son
diagnósticos, el nivel elevado de interleucina-6 y la linfadenopatía de volumen
pequeño con captación baja de FDG también son compatibles con iMCD.
Además, este paciente tiene características de un
subtipo clínico grave de iMCD llamado TAFRO, que se caracteriza por
trombocitopenia (T), anasarca (A), fiebre o un nivel elevado de proteína C
reactiva o ambos (F), insuficiencia renal o fibrosis de reticulina. o ambos
(R), y organomegalia (O). 13Aunque no se requiere para el diagnóstico,
hiperplasia megacariocítica en la médula ósea, mielofibrosis, características
hialinovasculares o hipervasculares en el tejido de los ganglios linfáticos, un
nivel elevado de fosfatasa alcalina, niveles bajos o normales de
gammaglobulinas y una masa mediastínica a menudo están presentes en pacientes
con iMCD –TAFRO. Los pacientes con iMCD que no cumplen con los criterios para
el subtipo TAFRO, que a menudo se describen con iMCD, no especificado de otra
manera (iMCD-NOS), tienden a tener trombocitosis, hipergammaglobulinemia y
características plasmocíticas en el tejido de los ganglios linfáticos. Aunque
la presentación clínica de este paciente es muy consistente con iMCD-TAFRO, se
requiere una revisión completa del tejido de los ganglios linfáticos para
determinar si los hallazgos histopatológicos respaldan un diagnóstico de iMCD
y, de ser así, para determinar qué variante histopatológica está presente.
Con respecto al diagnóstico de tumores mediastínicos,
un Case Record publicado en el Journal en 1964 señaló que "'cualquier
diagnóstico que no sea histológico en esta área es, en el mejor de los casos,
una conjetura fundamentada'; no estoy seguro de qué es en el fondo". el
peor." 14 Cuando también están presentes linfadenopatía y síntomas
sistémicos progresivos, es esencial una revisión histopatológica crítica de
tejido representativo adecuado.
DIAGNÓSTICO CLÍNICO PRESUNTIVO:
ENFERMEDAD DE CASTLEMAN MULTICÉNTRICA IDIOPÁTICA
(SUBTIPO TAFRO).
DISCUSIÓN PATOLÓGICA
Se obtuvieron muestras del segundo hospital para su
revisión. En la tinción con hematoxilina y eosina, la masa mediastínica
anterior mostró tejido fibroadiposo con fibrosis, necrosis grasa y
proliferación vascular benigna, todos ellos hallazgos inespecíficos. El tejido
del ganglio linfático de nivel VII ( Figura 2A y 2B) mostró distorsión
arquitectónica por una proliferación de folículos que tenían centros germinales
involucionados con focos ocasionales de hialinización. Varios centros
germinales fueron penetrados por vasos, lo que creó una apariencia de " lollipop".
Alrededor de los centros germinales involucionados había zonas de manto
prominentemente expandidas, que se caracterizaban por anillos concéntricos de
linfocitos con apariencia de “piel de cebolla”. El espacio interfolicular tenía
una vascularización aumentada, resaltada por una inmunotinción ERG, y también
tenía focos de células plasmáticas aumentadas, eosinófilos dispersos e
histiocitos antracóticos. La hibridación in situ para cadenas ligeras de
inmunoglobulina kappa y lambda mostró expresión politípica en las células plasmáticas.
Una tinción inmunohistoquímica para CD21 ( Figura 2C) mostró mallas de células
dendríticas foliculares asociadas con los folículos, con "gemelos"
ocasionales (dos centros germinales que comparten una sola malla de células
dendríticas foliculares). Una tinción inmunohistoquímica para HHV-8 fue
negativa. En el contexto de la presentación clínica de este paciente, estos
hallazgos son consistentes con la variante histopatológica hialino-vascular de
la enfermedad de Castleman.

Figura 2. Muestras de biopsia de tejido de ganglios
linfáticos y médula ósea.
El tejido de los ganglios linfáticos se obtuvo en el
segundo hospital en el momento de la cirugía torácica asistida por video. La
tinción con hematoxilina eosina (Paneles A y B) muestra un folículo que tiene
un centro germinal involucionado con un foco de hialinización y un vaso
penetrante (Panel A, flecha). La zona del manto circundante se expande y tiene
anillos concéntricos de linfocitos con aspecto de “piel de cebolla”. Hay un
aumento de la vascularización en el espacio interfolicular (Panel B), resaltado
por una inmunotinción ERG de los vasos (Panel B, recuadro). Una tinción inmunohistoquímica
para CD21 (Panel C) muestra folículos "gemelos" ocasionales, con dos
centros germinales (flechas) que comparten una sola malla de células dendríticas.
En este hospital se obtuvo una muestra de biopsia de médula ósea. Tinción con hematoxilina
y eosina (Panel D), muestra médula hipercelular con un marcado aumento en la
relación mieloide:eritroide. Hay un mayor número de megacariocitos (flechas),
que a menudo están presentes en pequeños grupos y tienen características
morfológicas normales.
Examen de una muestra de biopsia de médula ósea
obtenida en este hospital ( Figura 2D) reveló una médula hipercelular, con una
celularidad de la médula del 70%. No hubo evidencia de fibrosis de reticulina
patológicamente significativa. La relación mieloide:eritroide estaba
notablemente aumentada, y tanto los elementos mieloides como los eritroides
mostraron una maduración completa. Hubo un aumento en el número de
megacariocitos, que a menudo estaban presentes en pequeños grupos y tenían
características morfológicas normales. No hubo evidencia de agregados linfoides
o aumento de células plasmáticas. La citometría de flujo realizada en la
muestra de médula ósea no mostró evidencia de una población de células B
monoclonales o una población de células T inusual. Estos cambios son
consistentes con un proceso reactivo inespecífico, sin evidencia de una
enfermedad primaria de la médula ósea. La relación de los hallazgos de la
médula ósea con el diagnóstico de la enfermedad de Castleman es incierta,15 El
cuadro clínico es compatible con iMCD-TAFRO y se observaron ciertas
características histopatológicas, como las características hialino-vasculares
en el tejido de los ganglios linfáticos y el aumento del número de
megacariocitos en la médula ósea. Sin embargo, la fibrosis de reticulina en la
médula ósea, que se puede ver con iMCD-TAFRO, no estaba presente en este caso.
11
DIAGNÓSTICO PATOLÓGICO
VARIANTE HISTOPATOLÓGICA HIALINO-VASCULAR DE LA
ENFERMEDAD DE CASTLEMAN MULTICÉNTRICA IDIOPÁTICA.
DISCUSIÓN DEL MANEJO
Siltuximab, un anticuerpo monoclonal que se une a la
interleucina-6, ha sido aprobado por la Administración de Alimentos y
Medicamentos (FDA) para el tratamiento de pacientes con iMCD, y las pautas
actuales recomiendan siltuximab como terapia inicial. 16,17 En un ensayo aleatorizado,
doble ciego, controlado con placebo, de fase 2 que involucró a pacientes con
iMCD, el punto final primario de una respuesta radiográfica y sintomática
duradera a las 18 semanas ocurrió en 18 de 53 pacientes que recibieron
siltuximab (34 %) , pero el punto final primario no ocurrió en ninguno de los
18 pacientes que tenían la variante histopatológica hialino-vascular, de
acuerdo con criterios preespecificados evaluados en un laboratorio central de
patología. 18Siltuximab se asoció con tasas bajas de respuesta completa (2 %) y
respuesta sintomática completa (25 %) en el ensayo fundamental de fase 2, no se
ha comparado con un control activo (hasta donde sabemos) y requiere terapia
intravenosa continua cada 3 semanas hasta la progresión de la enfermedad o la
interrupción del tratamiento debido a efectos adversos.
Alternativamente, la quimioterapia citotóxica
combinada también es activa contra esta enfermedad y se ha asociado con
respuestas objetivas frecuentes entre pacientes con iMCD en series retrospectivas.
19,20 Dado que este paciente con iMCD presentaba insuficiencia orgánica, pensé
que se necesitaba una intervención confiablemente activa con un efecto
terapéutico rápido. Se procedió a la administración de R-CVP (rituximab,
ciclofosfamida, vincristina y prednisona) cada 3 semanas durante seis ciclos;
este régimen quimioterapéutico tiene un perfil de efectos secundarios aceptable
y está limitado en el tiempo. 21
Posteriormente, se demostró que siltuximab tiene un
beneficio en una pequeña serie de casos retrospectiva que involucró a pacientes
con la variante histopatológica hialina-vascular de iMCD y hallazgos
inflamatorios sistémicos, lo que llevó a algunos expertos a recomendar su uso
como terapia de primera línea en pacientes como este. . 22 Interpretaría estos
datos con cautela, dadas las limitaciones inherentes asociadas con un pequeño
estudio retrospectivo y dado que no se observaron respuestas objetivas entre
los pacientes con enfermedad vascular hialina en el ensayo pivotal de fase 2.
18 Siltuximab no es mi terapia de primera línea preferida para este paciente
con la variante histopatológica hialino-vascular de iMCD, pero es una opción si
este paciente tiene una recaída.
Se recomendó una evaluación de la respuesta
radiográfica y sintomática después de los primeros dos ciclos de tratamiento y
nuevamente al final del tratamiento. La evaluación incluyó anamnesis y examen
físico; evaluación de laboratorio para obtener el hemograma completo, perfil
químico sanguíneo y niveles de albúmina, citocinas y marcadores inflamatorios;
y seguimiento de imágenes PET-CT.
Este paciente completó seis ciclos de R-CVP sin
efectos adversos. Tuvo una respuesta rápida y sus síntomas relacionados con
iMCD, anasarca, hipoalbuminemia, lesión renal y pancitopenia se resolvieron en
días o semanas. Las exploraciones PET-CT obtenidas en el punto intermedio y al
final del tratamiento confirmaron la remisión completa. La revisión de las
imágenes en serie posteriores, así como las exploraciones de referencia,
sugirió una presentación previa al tratamiento de la enfermedad oligocéntrica
que afecta solo a los ganglios linfáticos mediastínicos, que está mal definida
y puede compartir características con la enfermedad de Castleman unicéntrica
(UCD) o iMCD. 23Se desconoce si la enfermedad de Castleman oligocéntrica se
trata mejor como UCD o como iMCD. Si bien la radioterapia generalmente no se
recomienda para pacientes con iMCD, es una opción de tratamiento con intención
curativa para pacientes con UCD que no pueden someterse a una resección quirúrgica
completa. 24-31 Por lo tanto, se pensó que este paciente con enfermedad
localizada radiográficamente podría beneficiarse de la radioterapia. Un equipo
multidisciplinario discutió las opciones de tratamiento y señaló que su
enfermedad podría incluirse de manera segura en un campo de radiación. Al
comprender el equilibrio de riesgos y beneficios potenciales, el paciente optó
por la radioterapia de consolidación. 24 Completó la terapia hace 3 años y
permanece sin evidencia de enfermedad.
PERSPECTIVA DEL COMENTARISTA
Aunque iMCD puede progresar rápidamente hasta
convertirse en una tormenta mortal de citoquinas, diagnosticar iMCD es un
desafío. Dada la urgencia diagnóstica, iMCD debe estar en el diagnóstico
diferencial cada vez que un síndrome inflamatorio y linfadenopatía se hayan
desarrollado simultáneamente o cada vez que se considere HLH ( Figura 3 ).


Figura 3. Condiciones caracterizadas por inflamación y
diagnóstico diferencial para iMCD.
El panel A muestra varias enfermedades autoinmunes o
relacionadas con el sistema inmunitario, condiciones iatrogénicas, malignas e
infecciosas que pueden causar una inflamación clínicamente significativa.
Algunas de estas condiciones puede progresar a una tormenta de citoquinas, que
se caracteriza por disfunciones orgánicas impulsadas por citoquinas. El panel B
muestra características y diagramas superpuestos para los dos subtipos de Enfermedad
de Castleman multicéntrica idiopática (iMCD): NOS (no especificado de otra
manera), y TAFRO (caracterizado por
trombocitopenia [T], anasarca [A], fiebre o nivel de proteína C reactiva
elevada o ambos [F], insuficiencia renal o fibrosis reticulina o ambos [R] y
organomegalia [O]). Pacientes con iMCD que no cumplen con los criterios para
iMCD–TAFRO son descritos como iMCD-NOS o iMCD, idiopáticos linfadenopatía plasmocítica
ic (iMCD-IPL). ALPES denota síndrome linfoproliferativo autoinmune, receptor de
antígeno quimérico CAR, citomegalovirus CMV, Covid-19 enfermedad por coronavirus
2019, CRS cytokine reléase syndrome, virus EBV Epstein-Barr, FDCS folicular sarcoma
de células dendríticas, virus de la hepatitis B HBV, HHV-8 herpesvirus humano 8,
inmunodeficiencia humana VIH virus, linfohistiocitosis hemofagocítica HLH, AIJ artritis
idiopática juvenil, activación de macrófagos MAS, enfermedad de Castleman
multicéntrica MCD, Linfoma hemofagocítico asociado a malignidad M-HLH linfohistiocitosis,
polineuropatía POEMS, organomegalia, endocrinopatía, proteína M y cambios en la
piel, LES lupus eritematoso sistémico y TB tuberculosis
Me alegra saber lo bien que le está yendo a este paciente,
gracias a sus astutos médicos y los avances en el diagnóstico y tratamiento de
la enfermedad de Castleman desde la descripción original de Benjamin Castleman
en un Case Record publicado en el Journal en 1954 32— particularmente durante
los últimos 12 años, desde el momento en que me diagnosticaron iMCD, en 2010.
Gracias a los esfuerzos internacionales durante la última década, ahora tenemos
las primeras pautas diagnósticas de consenso basadas en evidencia 12 y pautas
de tratamiento, 16 las primeras descripciones de iMCD–TAFRO, 33 el primer
tratamiento aprobado por la FDA, 18 el primer registro de pacientes (Número de
ClinicalTrials.gov, NCT02817997. se abre en una pestaña nueva), 34 y una mejora
en la supervivencia general a 5 años del 65 % en 2012 al 75 % en 2021. 35,36
DIAGNOSTICO FINAL
ENFERMEDAD DE CASTLEMAN MULTICÉNTRICA IDIOPÁTICA.
Fuente:
David C. Fajgenbaum, M.D., Reece J. Goiffon, M.D.,
Ph.D., Jacob D. Soumerai, M.D., and Cynthia K. Harris, M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2115849
Referencias.
1. Fajgenbaum DC, June CH. Cytokine
storm. N Engl J Med 2020;383:2255-73.
2. Küppers R. Molecular biology of
Hodgkin’s lymphoma. Adv Cancer Res
2002;84:277-312.
3. Seriburi V, Ndukwe N, Chang Z, Cox
ME, Wormser GP. High frequency of false
positive IgM immunoblots for Borrelia
burgdorferi in clinical practice. Clin Mi[1]crobiol
Infect 2012;18:1236-40.
4. Kalish RA, McHugh G, Granquist J,
Shea B, Ruthazer R, Steere AC. Persis[1]tence
of immunoglobulin M or immuno[1]globulin
G antibody responses to Borrelia
burgdorferi 10-20 years after active Lyme
disease. Clin Infect Dis 2001;33:780-5.
5. Slater CA, Davis RB, Shmerling RH.
Antinuclear antibody testing: a study of
clinical utility. Arch Intern Med 1996;156:
1421-5.
6. Liang E, Taylor M, McMahon M. Utility
of the AVISE Connective Tissue Disease
test in predicting lupus diagnosis and
progression. Lupus Sci Med 2020;7(1):
e000345.
7. Barton JC, Barton JC, Bertoli LF, Ac[1]ton
RT. Characterization of adult pa[1]tients
with IgG subclass deficiency and
subnormal IgG2. PLoS One 2020;15(10):
e0240522.
8. Allen CE, Yu X, Kozinetz CA, McClain
KL. Highly elevated ferritin levels and the
diagnosis of hemophagocytic lymphohis[1]tiocytosis.
Pediatr Blood Cancer 2008;50:
1227-35.
9. Jordan MB, Allen CE, Greenberg J, et al.
Challenges in the diagnosis of hemo[1]phagocytic
lymphohistiocytosis: recom[1]mendations
from the North American
Consortium for Histiocytosis (NACHO).
Pediatr Blood Cancer 2019;66(11):e27929.
10. Hayden A, Lin M, Park S, et al. Soluble
interleukin-2 receptor is a sensitive diag[1]nostic
test in adult HLH. Blood Adv 2017;
1:2529-34.
11. Dispenzieri A, Fajgenbaum DC. Over[1]view
of Castleman disease. Blood 2020;
135:1353-64.
12. Fajgenbaum DC, Uldrick TS, Bagg A,
et al. International, evidence-based con[1]sensus
diagnostic criteria for HHV-8-neg[1]ative/idiopathic
multicentric Castleman
disease. Blood 2017;129:1646-57.
13. Nishimura Y, Fajgenbaum DC, Pier[1]son
SK, et al. Validated international defi[1]nition
of the thrombocytopenia, anasarca,
fever, reticulin fibrosis, renal insuffi[1]ciency,
and organomegaly clinical sub[1]type
(TAFRO) of idiopathic multicentric
Castleman disease. Am J Hematol 2021;
96:1241-52.
14. Case Records of the Massachusetts
General Hospital (Case 57-1964). N Engl J
Med 1964;271:1160-5.
15. Belyaeva E, Rubenstein A, Pierson SK,
et al. Bone marrow findings of idiopathic
Multicentric Castleman disease: a histo[1]pathologic
analysis and systematic litera[1]ture
review. Hematol Oncol 2022;40:191-
201.
16. van Rhee F, Voorhees P, Dispenzieri A,
et al. International, evidence-based con[1]sensus
treatment guidelines for idiopath[1]ic
multicentric Castleman disease. Blood
2018;132:2115-24.
17. Zelenetz AD, Gordon LI, Chang JE,
et al. NCCN guidelines insights: B-cell
lymphomas, version 5.2021. J Natl Compr
Canc Netw 2021;19:1218-30.
18. van Rhee F, Wong RS, Munshi N, et al.
Siltuximab for multicentric Castleman’s
disease: a randomised, double-blind, pla[1]cebo-controlled
trial. Lancet Oncol 2014;
15:966-74.
19. Dong Y, Zhang L, Nong L, et al. Effec[1]tiveness
of rituximab-containing treat[1]ment
regimens in idiopathic multicentric
Castleman disease. Ann Hematol 2018;97:
1641-7.
20. Liu AY, Nabel CS, Finkelman BS, et al.
Idiopathic multicentric Castleman’s dis[1]ease:
a systematic literature review. Lan[1]cet
Haematol 2016;3(4):e163-e175.
21. Luminari S, Ferrari A, Manni M, et al.
Long-term results of the FOLL05 trial
comparing R-CVP versus R-CHOP versus
R-FM for the initial treatment of patients
with advanced-stage symptomatic follic[1]ular
lymphoma. J Clin Oncol 2018;36:689-
96.
22. Fajgenbaum DC, Wu D, Goodman A,
et al. Insufficient evidence exists to use
histopathologic subtype to guide treatment
of idiopathic multicentric Castleman dis[1]ease.
Am J Hematol 2020;95:1553-61.
23. van Rhee F, Oksenhendler E, Srkalovic
G, et al. International evidence-based con[1]sensus
diagnostic and treatment guide[1]lines
for unicentric Castleman disease.
Blood Adv 2020;4:6039-50.
24. Beckham TH, Yang JC, Chau KW, Noy
A, Yahalom J. Excellent outcomes with
surgery or radiotherapy in the manage[1]ment
of castleman disease including a
case of oligocentric disease. Clin Lym[1]phoma
Myeloma Leuk 2020;20:685-9.
25. Chronowski GM, Ha CS, Wilder RB,
Cabanillas F, Manning J, Cox JD. Treat[1]ment
of unicentric and multicentric
Castleman disease and the role of radio[1]therapy.
Cancer 2001;92:670-6.
26. de Vries IA, van Acht MMS, Demeyere
TBJ, Lybeert ML, de Zoete J-P, Nieuwen[1]huijzen
GA. Neoadjuvant radiotherapy of
primary irresectable unicentric Castle[1]man’s
disease: a case report and review of
the literature. Radiat Oncol 2010;5:7.
27. Li YM, Liu PH, Zhang YH, et al. Radio[1]therapy
of unicentric mediastinal Castle[1]man’s
disease. Chin J Cancer 2011;30:
351-6.
28. Matthiesen C, Ramgopol R, Seavey J,
Ahmad S, Herman T. Intensity modulated
radiation therapy (IMRT) for the treat[1]ment
of unicentric Castlemans disease:
a case report and review of the use of ra[1]diotherapy
in the literature. Radiol Oncol
2012;46:265-70.
29. Miranda FA, Faria VHC, Arruda GV,
Silva LG. Radiation therapy in the treat[1]ment
of unicentric Castleman’s disease.
J Bras Pneumol 2013;39:116-8.
30. Neuhof D, Debus J. Outcome and late
complications of radiotherapy in patients
with unicentric Castleman disease. Acta
Oncol 2006;45:1126-31.
31. Noh OK, Lee S-W, Lee JW, et al. Cases
report of unicentric Castleman’s disease:
revisit of radiotherapy role. Radiat Oncol J
2013;31:48-54.
32. Case Records of the Massachusetts
General Hospital (Case 40011). N Engl J
Med 1954;250:26-30.
33. Iwaki N, Fajgenbaum DC, Nabel CS,
et al. Clinicopathologic analysis of TAFRO
syndrome demonstrates a distinct sub[1]type
of HHV-8-negative multicentric
Castleman disease. Am J Hematol 2016;
91:220-6.
34. Pierson SK, Khor JS, Ziglar J, et al.
ACCELERATE: a patient-powered natural
history study design enabling clinical and
therapeutic discoveries in a rare disorder.
Cell Rep Med 2020;1:100158.
35. Dispenzieri A, Armitage JO, Loe MJ,
et al. The clinical spectrum of Castleman’s
disease. Am J Hematol 2012;87:997-1002.
36. Cohen AB, Swaminathan A, Wang X,
et al. Clinical characteristics, treatment
patterns, and overall survival of real[1]world
patients with idiopathic multicen[1]tric
Castleman disease. In: Proceedings
and abstracts of the 2021 Annual Meeting
of the American Society of Clinical Onco
No hay comentarios:
Publicar un comentario