
Paciente de sexo femenino de 69 años que se interna por presentar tumoración de mama izquierda de aproximadamente 8 cm de diámetro, dura, movil no adherida a planos profundos, no dolorosa. “Según refiere la paciente de 20 dias de evolución”. Acompañada de decaimiento y palidez.
Al examen físico no presenta adenopatías axilares ni supraclaviculares.
Antecedentes personales: HTA en tratamiento, refiere haber presentado un episodio de hematuria que cedió espontáneamente. Neumonía en año 2000. No cirugías. Pérdida de peso en el ultimo mes (se desconoce cuanto).
Laboratorio:G.B: 4.300 ( N:70 %. L:24 %), G.R: 3.590.000Hb: 6,4 gr/dl.Hto: 21,8 % VCM 60,7, Plaq:388.000 mm3.Glucemia : 111 mg% VSG: 120 mm, Uricemia: 4,30 Ionograma: Na: 137 mmol/L, K: 3,9 mmol/L, CL: 106 mmol/L, TGO: 23 UI / L, TGP:11 UI / L, FAL: 160 UI / L, Bil. D: 0,10 mg %, Bil. I: 0,60 mg %, Bil. T: 0,70 mg Amilasa: 45 UI / L
GASES: pH: 7,42, pCO2: 28 mmHg., pO2: 146 mmHg., HCO3: 18,1 mmol / L, BE: - 6,2mmol / L, Sat. O2: 99 %
1º Día de Internación: Pte lúcida, afebril. Palidez cutáneo- mucosa. Presenta mama izq. indurada, con aumento del tamaño y circulación periférica colateral. No presenta adenopatías periféricas. Abdomen: sp.
Varices en Miembros Inferiores. Resto del Ex. F: s/p
Examen de la mama izquierda: mama mediana, péndula ,pezón y areola Se palpa en Mama Izq. Tumoración de 8 x 10 cm. aprox, móvil, continuo, indurada. : s/p.Mama Der: s/p.
Piel: s/p.Región Axilar: ( - )Región Supraclavicular: ( - )
Mamografía: Densidades Asimétricas redondeadas con calcificaciones en su interior en mama Der. compatible con Fibroadenomas calcificados.
Mama Izq: Con importante aumento de su densidad generalizado, donde impresiona existir además densidades asimétricas redondeadas de bordes definidos.
Conclusión: BI – RADS ???
ECO Mamaria: Mama Der: s/ p: Mama Izq: con imagen de aspecto sólido, de bordes netos y lobulados. Ecoestructura heterogénea en forma difusa.
2º Día de Internación: ECO Hepática: Higado de tamaño y ecoestructura normal sin imágenes focales ecograficamente detectables.
Se realiza ( PAAF ) Punción Aspiración con aguja Fina.
Se transfunde con 2 U de G.R.
3º Día de Internación: Diagnóstico Presuntivo:
Tumor Phylloides de mama
Lab: G.B: 4.400 ( N:66 %. L:26 %)Hb: 6,4 gr/dl. Hto: 22 %Plaq:349.000 mm3.
Glu: 78 mg%, Creatinina: 1 mg%, Urea: 33 mg %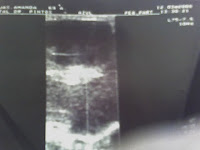
Ionograma:Na: 138 mmol/L, K: 4,06 mmol/L, CL: 105 mmol/L
TGO: 20 UI / L, TGP:10 UI / L, FAL: 141 UI / L, Bil. D: 0,12 mg %, Bil. I: 0,57 mg %, Bil. T: 0,69 mg %, Col. T: 133 mg%,
Coagulograma: Tiempo y [ ] de Protrombina: 13 seg. 100 %
Informe de PAAF:Macroscopía: 2 extendidos.Diagnóstico: Colgajos Epiteliales congestivos y escasos Linfocitos. Consistente con Tumor Phyllodes.
6º Día de Internación: SOMF ( no se logro realizar)
Se transfunde 1 U de G.R.( pre- cirugia)
7º Día de Internación. Se realiza Cirugía Mamaria Izquierda: Tumorectomía de Mama: tumor de 10 x 10 cm.
8 º Día de Internación: ECO Ginecológica, Renal y Vesical:
Riñones: s /p.Vejiga: de paredes lisas, con imágen de aspecto sólida en desembocadura del Uréter Derecho de aproximadamente 1,7 cm compatible con Pólipo y / o Coágulo.Útero y Anexos: s /p.
Se realiza IC con Servicio de Urología. IC de Urología: Se indica realizar Urograma Excretor para poder descartar Tumor de Vejiga ya que tiene imagen en desembocadura de uréter derecho.
10 º Día de Internación: 3 º día de posoperatorio. Buena evolución. Pte sin dolor. Herida de bordes afrontados, seca, sin signos de flogosis.
Alta Institucional con seguimiento Clínico, Urológico y Ginecológico
Resultado de Anatomía Patológica
Macroscopía: Se recibe resección mamaria que mide 10 cm de diámetro mayor, consistente en tumoración polilobulada, blanda, homogénea, amarillenta. Se muestrea según técnica.
Microscopía: Los cortes histológicos muestran proliferación Linfomatosa Atípica Difusa No Hodgkin compuestas por Células de mediano tamaño algunas con nucleolo prominentes.
Diagnóstico: Linfoma mamario No Hodgkin Difuso Tipo B de células intermedias.
Diagnóstico: Linfoma Primario? de mama
Discusión: Se presentó una paciente de 69 años, con tumoración de 10 cm dura de rápido crecimiento, que al decir de la paciente tenía 20 días de evolución???.
Además, la paciente presentaba un compromiso general importante con adelgazamiento significativo, aunque no cuantificado y con un síndrome anémico severo.
La paciente es llevada rápidamente a cirugía donde se le realiza tumorectomía, con diagnóstico presuntivo preoperatorio, por elementos clínicos y PAAF de Tumor Phylloides de mama. Se consideró el diagnóstico de tumor Phylloides de mama, por lo que se desprende de la historia clínica, al dar crédito a la apreciación de la paciente de el extremadamente rápido crecimiento. Este tumor benigno de la mama , también conocido como Fibroadenoma intracanalicular, catalogado como una variante del Fibroadenoma, tiene dos características: crecimiento rápido y gran tamaño, y debe ser diferenciado de los sarcomas
Etiológicamente se acepta su origen primitivo que representaría el 2-3% de los fibroadenomas o puede venir de la transformación de un fibroadenoma que presentó en su inicio zonas mixoides. Representa el 0.5%-2% de los tumores de la mama y clínicamente se presenta antes de los 25 años siendo confundido con un fibroadenoma juvenil o, más frecuentemente, después de los 40 años. En la mayoría de las ocasiones aparece como una masa bien definida, unilateral, indolora, móvil, que se caracteriza por un crecimiento rápido y puede afectar a la piel por compresión, adquiriendo un tamaño grande que puede superar los 10 cm., pero también se han descrito t. phyllodes de 1 cm.
Su crecimiento puede presentar dos formas, una forma bifásica en la que en una primera etapa casi no se percibe durante un periodo largo, y en una segunda fase, más corta como de unos 7 meses, presenta un crecimiento rápido. La otra forma evolutiva es monofásica con crecimiento lento (en años) o rápido (en meses).
La histología, demuestra un importante crecimiento de los tejidos epitelial y estromal.
Macroscópicamente son tumores grandes que pueden alcanzar los 10 cm, de aspecto lobulado, parduzco, bien delimitado, de consistencia firme y blanda, con cápsula. Al microscopio es un tumor mixto, epitelial y conjuntivo, en el que el componente epitelial es ductal y mioepitelial sin atipia ni alteraciones, pero el estroma predomina sobre el epitelio, siendo un estroma denso de tipo fibroblástico con actividad mitótica, cuyo crecimiento da lugar a nodulaciones de estroma cubiertas por epitelio. Aparentemente se origina en el estroma intralobulillar y periductal, sensible a la acción hormonal. Su caracter benigno queda definido por la escasa atipia celular y necrosis, con menos de 5 mitosis por campo de gran aumento, y ausencia de carácter infiltrante en la perifería.En los casos malignos tiene un mínimo potencial de metastatización a los ganglios linfáticos regionales.
Sea benigno o maligno casi nunca es multifocal. En función del tumor marginal, crecimiento del componente conectivo, mitosis en el área de mayor actividad del tumor, celularidad del estroma y atipias celulares, el tumor phyllodes puede ser clasificado en benigno, borderline y maligno.El tratamiento es la resección, nunca la enucleación, con un margen de 1-2 cm. para evitar las recidivas (tumorectomía ampliada), con control histológico intraoperatorio de los bordes quirúrgicos, que en el caso de que apareciesen y si el resto de la glándula lo permiten también serían extirpadas. Si el tejido mamario restante es escaso puede ser necesaria para su extirpación la realización de una mastectomía subcutánea con la colocación de prótesis.No precisa linfadenectomia aunque el diagnóstico histológico confirme componente sarcomatoso, ya que la afectación ganglionar no supera el 2% de los casos.
Se consideró durante el ateneo que el diagnóstico presuntivo preoperatorio conspiró contra lo que debiera ser la sistemática de cualquier tumor de mama que es realizar una biopsia por congelación intraoperatoria par tomar la conducta adecuada de acuerdo a la misma. De haberse realizado en este caso la cirugía hubiese sido mas amplia (mastectomía parcial o total), pero con exploración de la axila.
En el análisis del caso son evidentes algunas inconsistencias en el estudio de la paciente, ya que un tumor benigno de mama no debiera presentar un compromiso importante del estado general como el de esta paciente, así como la presencia de anemia significativa (Hematocrito de 21%) y VSG de 120 mm?
Con respecto a la anemia se consideró en el ateneo que fue incompletamente estudiada. Se trataba por lo que se desprende de los índices hematimétricos de una anemia microcítica (VCM 60,7 mm3), que debiera haber despertado la sospecha de un déficit de hierro como primer diagnóstico, no llevándose a cabo preoperatoriamente estudios del metabolismo del hierro (Transferrina, Ferremia, Saturación de Transferrina, Ferritina plasmática etc), así como tampoco una investigación de SOMF. La paciente tenía hematuria? que podría haber sido una causa de anemia ferropénica, aunque por lo que se desprende de la historia clínica, no se realizó nunca un sedimento de orina para buscar hematíes en el mismo. De haber tenido una hematuria significativa y repetida podría justificar anemia ferropénica como es común de ver en cáncer de vejiga riñón etc. Si en cambio en vez de hematuria se hubiese encontrado en el sedimento solamente hemoglobina y no glóbulos rojos, la Hemoglobinuria Paroxística Benigna hubiese sido otra consideración.
No figuran en la historia clínica valore sde reticulocitos que podrían haber ayudado a comprender la patogénesis de la anemia y haberla clasificado como hiper o hiporregenerativa. No figuran tampoco valores de LDH que podrían haber sido necesarios no solo para descartar algún componente hemolítico de la anemia, sino también en el contexto del estudio de cualquier paciente con cáncer, y sobre todo si se está considerando la posibilidad de linfoma
Con respecto al diagnóstico definitivo de Linfoma Primario de mama se consideró que la paciente, además de no tener estudiado el inmunofenotipo del tumor, tampoco fue debidamente estatificada con TAC de tórax, abdomen y pelvis, así como tampoco con un estudio de Médula ósea (PAMO), y eventual biopsia ósea, por lo que sin estos elementos es imposible rotular de Primario al tumor en cuestión.

Presentó la Dra Andrea Suarez
Al examen físico no presenta adenopatías axilares ni supraclaviculares.
Antecedentes personales: HTA en tratamiento, refiere haber presentado un episodio de hematuria que cedió espontáneamente. Neumonía en año 2000. No cirugías. Pérdida de peso en el ultimo mes (se desconoce cuanto).
Laboratorio:G.B: 4.300 ( N:70 %. L:24 %), G.R: 3.590.000Hb: 6,4 gr/dl.Hto: 21,8 % VCM 60,7, Plaq:388.000 mm3.Glucemia : 111 mg% VSG: 120 mm, Uricemia: 4,30 Ionograma: Na: 137 mmol/L, K: 3,9 mmol/L, CL: 106 mmol/L, TGO: 23 UI / L, TGP:11 UI / L, FAL: 160 UI / L, Bil. D: 0,10 mg %, Bil. I: 0,60 mg %, Bil. T: 0,70 mg Amilasa: 45 UI / L
GASES: pH: 7,42, pCO2: 28 mmHg., pO2: 146 mmHg., HCO3: 18,1 mmol / L, BE: - 6,2mmol / L, Sat. O2: 99 %

1º Día de Internación: Pte lúcida, afebril. Palidez cutáneo- mucosa. Presenta mama izq. indurada, con aumento del tamaño y circulación periférica colateral. No presenta adenopatías periféricas. Abdomen: sp.
Varices en Miembros Inferiores. Resto del Ex. F: s/p
Examen de la mama izquierda: mama mediana, péndula ,pezón y areola Se palpa en Mama Izq. Tumoración de 8 x 10 cm. aprox, móvil, continuo, indurada. : s/p.Mama Der: s/p.
Piel: s/p.Región Axilar: ( - )Región Supraclavicular: ( - )
Mamografía: Densidades Asimétricas redondeadas con calcificaciones en su interior en mama Der. compatible con Fibroadenomas calcificados.
Mama Izq: Con importante aumento de su densidad generalizado, donde impresiona existir además densidades asimétricas redondeadas de bordes definidos.

Conclusión: BI – RADS ???
ECO Mamaria: Mama Der: s/ p: Mama Izq: con imagen de aspecto sólido, de bordes netos y lobulados. Ecoestructura heterogénea en forma difusa.
2º Día de Internación: ECO Hepática: Higado de tamaño y ecoestructura normal sin imágenes focales ecograficamente detectables.
Se realiza ( PAAF ) Punción Aspiración con aguja Fina.
Se transfunde con 2 U de G.R.
3º Día de Internación: Diagnóstico Presuntivo:
Tumor Phylloides de mama
Lab: G.B: 4.400 ( N:66 %. L:26 %)Hb: 6,4 gr/dl. Hto: 22 %Plaq:349.000 mm3.
Glu: 78 mg%, Creatinina: 1 mg%, Urea: 33 mg %

Ionograma:Na: 138 mmol/L, K: 4,06 mmol/L, CL: 105 mmol/L
TGO: 20 UI / L, TGP:10 UI / L, FAL: 141 UI / L, Bil. D: 0,12 mg %, Bil. I: 0,57 mg %, Bil. T: 0,69 mg %, Col. T: 133 mg%,
Coagulograma: Tiempo y [ ] de Protrombina: 13 seg. 100 %
Informe de PAAF:Macroscopía: 2 extendidos.Diagnóstico: Colgajos Epiteliales congestivos y escasos Linfocitos. Consistente con Tumor Phyllodes.
6º Día de Internación: SOMF ( no se logro realizar)
Se transfunde 1 U de G.R.( pre- cirugia)
7º Día de Internación. Se realiza Cirugía Mamaria Izquierda: Tumorectomía de Mama: tumor de 10 x 10 cm.
8 º Día de Internación: ECO Ginecológica, Renal y Vesical:
Riñones: s /p.Vejiga: de paredes lisas, con imágen de aspecto sólida en desembocadura del Uréter Derecho de aproximadamente 1,7 cm compatible con Pólipo y / o Coágulo.Útero y Anexos: s /p.
Se realiza IC con Servicio de Urología. IC de Urología: Se indica realizar Urograma Excretor para poder descartar Tumor de Vejiga ya que tiene imagen en desembocadura de uréter derecho.
10 º Día de Internación: 3 º día de posoperatorio. Buena evolución. Pte sin dolor. Herida de bordes afrontados, seca, sin signos de flogosis.
Alta Institucional con seguimiento Clínico, Urológico y Ginecológico
Resultado de Anatomía Patológica
Macroscopía: Se recibe resección mamaria que mide 10 cm de diámetro mayor, consistente en tumoración polilobulada, blanda, homogénea, amarillenta. Se muestrea según técnica.
Microscopía: Los cortes histológicos muestran proliferación Linfomatosa Atípica Difusa No Hodgkin compuestas por Células de mediano tamaño algunas con nucleolo prominentes.
Diagnóstico: Linfoma mamario No Hodgkin Difuso Tipo B de células intermedias.
Diagnóstico: Linfoma Primario? de mama
Discusión: Se presentó una paciente de 69 años, con tumoración de 10 cm dura de rápido crecimiento, que al decir de la paciente tenía 20 días de evolución???.
Además, la paciente presentaba un compromiso general importante con adelgazamiento significativo, aunque no cuantificado y con un síndrome anémico severo.
La paciente es llevada rápidamente a cirugía donde se le realiza tumorectomía, con diagnóstico presuntivo preoperatorio, por elementos clínicos y PAAF de Tumor Phylloides de mama. Se consideró el diagnóstico de tumor Phylloides de mama, por lo que se desprende de la historia clínica, al dar crédito a la apreciación de la paciente de el extremadamente rápido crecimiento. Este tumor benigno de la mama , también conocido como Fibroadenoma intracanalicular, catalogado como una variante del Fibroadenoma, tiene dos características: crecimiento rápido y gran tamaño, y debe ser diferenciado de los sarcomas
Etiológicamente se acepta su origen primitivo que representaría el 2-3% de los fibroadenomas o puede venir de la transformación de un fibroadenoma que presentó en su inicio zonas mixoides. Representa el 0.5%-2% de los tumores de la mama y clínicamente se presenta antes de los 25 años siendo confundido con un fibroadenoma juvenil o, más frecuentemente, después de los 40 años. En la mayoría de las ocasiones aparece como una masa bien definida, unilateral, indolora, móvil, que se caracteriza por un crecimiento rápido y puede afectar a la piel por compresión, adquiriendo un tamaño grande que puede superar los 10 cm., pero también se han descrito t. phyllodes de 1 cm.
Su crecimiento puede presentar dos formas, una forma bifásica en la que en una primera etapa casi no se percibe durante un periodo largo, y en una segunda fase, más corta como de unos 7 meses, presenta un crecimiento rápido. La otra forma evolutiva es monofásica con crecimiento lento (en años) o rápido (en meses).
La histología, demuestra un importante crecimiento de los tejidos epitelial y estromal.
Macroscópicamente son tumores grandes que pueden alcanzar los 10 cm, de aspecto lobulado, parduzco, bien delimitado, de consistencia firme y blanda, con cápsula. Al microscopio es un tumor mixto, epitelial y conjuntivo, en el que el componente epitelial es ductal y mioepitelial sin atipia ni alteraciones, pero el estroma predomina sobre el epitelio, siendo un estroma denso de tipo fibroblástico con actividad mitótica, cuyo crecimiento da lugar a nodulaciones de estroma cubiertas por epitelio. Aparentemente se origina en el estroma intralobulillar y periductal, sensible a la acción hormonal. Su caracter benigno queda definido por la escasa atipia celular y necrosis, con menos de 5 mitosis por campo de gran aumento, y ausencia de carácter infiltrante en la perifería.En los casos malignos tiene un mínimo potencial de metastatización a los ganglios linfáticos regionales.
Sea benigno o maligno casi nunca es multifocal. En función del tumor marginal, crecimiento del componente conectivo, mitosis en el área de mayor actividad del tumor, celularidad del estroma y atipias celulares, el tumor phyllodes puede ser clasificado en benigno, borderline y maligno.El tratamiento es la resección, nunca la enucleación, con un margen de 1-2 cm. para evitar las recidivas (tumorectomía ampliada), con control histológico intraoperatorio de los bordes quirúrgicos, que en el caso de que apareciesen y si el resto de la glándula lo permiten también serían extirpadas. Si el tejido mamario restante es escaso puede ser necesaria para su extirpación la realización de una mastectomía subcutánea con la colocación de prótesis.No precisa linfadenectomia aunque el diagnóstico histológico confirme componente sarcomatoso, ya que la afectación ganglionar no supera el 2% de los casos.
Se consideró durante el ateneo que el diagnóstico presuntivo preoperatorio conspiró contra lo que debiera ser la sistemática de cualquier tumor de mama que es realizar una biopsia por congelación intraoperatoria par tomar la conducta adecuada de acuerdo a la misma. De haberse realizado en este caso la cirugía hubiese sido mas amplia (mastectomía parcial o total), pero con exploración de la axila.
En el análisis del caso son evidentes algunas inconsistencias en el estudio de la paciente, ya que un tumor benigno de mama no debiera presentar un compromiso importante del estado general como el de esta paciente, así como la presencia de anemia significativa (Hematocrito de 21%) y VSG de 120 mm?
Con respecto a la anemia se consideró en el ateneo que fue incompletamente estudiada. Se trataba por lo que se desprende de los índices hematimétricos de una anemia microcítica (VCM 60,7 mm3), que debiera haber despertado la sospecha de un déficit de hierro como primer diagnóstico, no llevándose a cabo preoperatoriamente estudios del metabolismo del hierro (Transferrina, Ferremia, Saturación de Transferrina, Ferritina plasmática etc), así como tampoco una investigación de SOMF. La paciente tenía hematuria? que podría haber sido una causa de anemia ferropénica, aunque por lo que se desprende de la historia clínica, no se realizó nunca un sedimento de orina para buscar hematíes en el mismo. De haber tenido una hematuria significativa y repetida podría justificar anemia ferropénica como es común de ver en cáncer de vejiga riñón etc. Si en cambio en vez de hematuria se hubiese encontrado en el sedimento solamente hemoglobina y no glóbulos rojos, la Hemoglobinuria Paroxística Benigna hubiese sido otra consideración.
No figuran en la historia clínica valore sde reticulocitos que podrían haber ayudado a comprender la patogénesis de la anemia y haberla clasificado como hiper o hiporregenerativa. No figuran tampoco valores de LDH que podrían haber sido necesarios no solo para descartar algún componente hemolítico de la anemia, sino también en el contexto del estudio de cualquier paciente con cáncer, y sobre todo si se está considerando la posibilidad de linfoma
Con respecto al diagnóstico definitivo de Linfoma Primario de mama se consideró que la paciente, además de no tener estudiado el inmunofenotipo del tumor, tampoco fue debidamente estatificada con TAC de tórax, abdomen y pelvis, así como tampoco con un estudio de Médula ósea (PAMO), y eventual biopsia ósea, por lo que sin estos elementos es imposible rotular de Primario al tumor en cuestión.

Presentó la Dra Andrea Suarez





 Paciente de 65 años que consulta por síndrome de repercusión general, adelgazamiento de 15 kg en los últimos 4 meses acompañados de equivalentes febriles (escalofríos, sudoración), aunque nunca se ha controlado la temperatura. Tiene antecedentes de tabaquismo y etilismo intensos. Ha realizado trabajos en el campo, aunque actualmente está desocupado y en estado de abandono. En el examen físico llama la atención el intenso adelgazamiento. En el examen del abdomen llama la atención una hepatomegalia de 6 traveses de dedo por debajo del reborde costal, lisa e indolora.
Paciente de 65 años que consulta por síndrome de repercusión general, adelgazamiento de 15 kg en los últimos 4 meses acompañados de equivalentes febriles (escalofríos, sudoración), aunque nunca se ha controlado la temperatura. Tiene antecedentes de tabaquismo y etilismo intensos. Ha realizado trabajos en el campo, aunque actualmente está desocupado y en estado de abandono. En el examen físico llama la atención el intenso adelgazamiento. En el examen del abdomen llama la atención una hepatomegalia de 6 traveses de dedo por debajo del reborde costal, lisa e indolora.


























