El calcio es vital para la regulación de una amplia gama de procesos fisiológicos humanos, incluida la división celular, la adhesión celular, la integridad de la membrana plasmática, la secreción de proteínas y hormonas, la contracción muscular, la excitabilidad neuronal, el metabolismo del glucógeno, la agregación plaquetaria y la coagulación sanguínea. Dada esta lista, no sorprende que los niveles de calcio estén muy estrictamente regulados.
El cuerpo humano contiene alrededor de 1000 g de
calcio, de los cuales >99% reside en el esqueleto. Esto deja <1% del
calcio corporal total en la fase soluble, dividido entre los compartimentos líquidos
intracelular y extracelular. La concentración de calcio extracelular es
aproximadamente 10 000 veces mayor que la concentración de calcio intracelular,
pero ambos son fundamentales para el correcto funcionamiento de los procesos
fisiológicos. Del calcio extracelular, el 50% está unido (40% a albúmina; 10% a
citrato, fosfato y otros iones), mientras que el otro 50% está libre o
ionizado. Solo el calcio ionizado es biológicamente activo y solo esta fracción
está regulada hormonalmente. Para ajustar el nivel de calcio para las
elevaciones de las proteínas plasmáticas, el calcio sérico total debe reducirse
en 0,8 mg/dL por cada 1 g/dL de albúmina por encima del rango normal.
La exquisita regulación del calcio ionizado es
verdaderamente notable, dado el suministro siempre cambiante de calcio de la
dieta frente a las demandas constantes de calcio por parte de varios tejidos en
todo el cuerpo. Dada la complejidad del sistema, no sorprende que se necesiten
cuatro órganos diferentes y al menos dos hormonas diferentes para mantener la
homeostasis del calcio alrededor de un punto de ajuste deseado. Los cuatro
órganos clave son las paratiroides, el intestino, los riñones y el esqueleto,
mientras que las hormonas reguladoras más críticas son la hormona paratiroidea
(PTH) y la vitamina D. El magnesio y el fósforo, así como la calcitonina y el
factor de crecimiento de fibroblastos 23, contribuyen a la homeostasis mineral.
Cuando el calcio sérico aumenta entre un 2 % y un 3 %
por encima del punto de ajuste determinado genéticamente, los mecanismos
homeostáticos se activan rápidamente para que el nivel vuelva a la normalidad.
En primer lugar, en las glándulas paratiroides, el exceso de calcio actúa a
través de innumerables receptores sensores de calcio (CaSR) para detener
inmediatamente la secreción de PTH. Los CaSR son receptores transmembrana
acoplados a proteína G que son sumamente sensibles a los cambios en la
concentración de calcio ionizado (fig. 51.1).
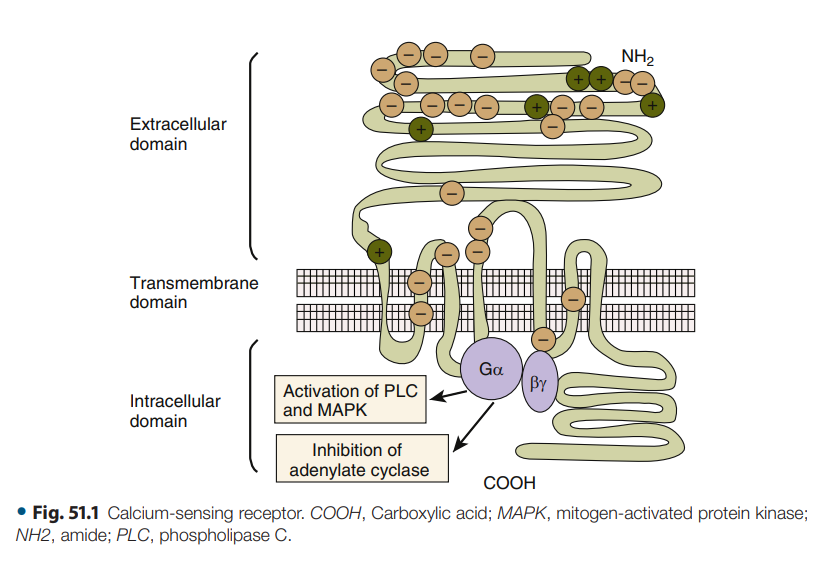
Figura 51.1.
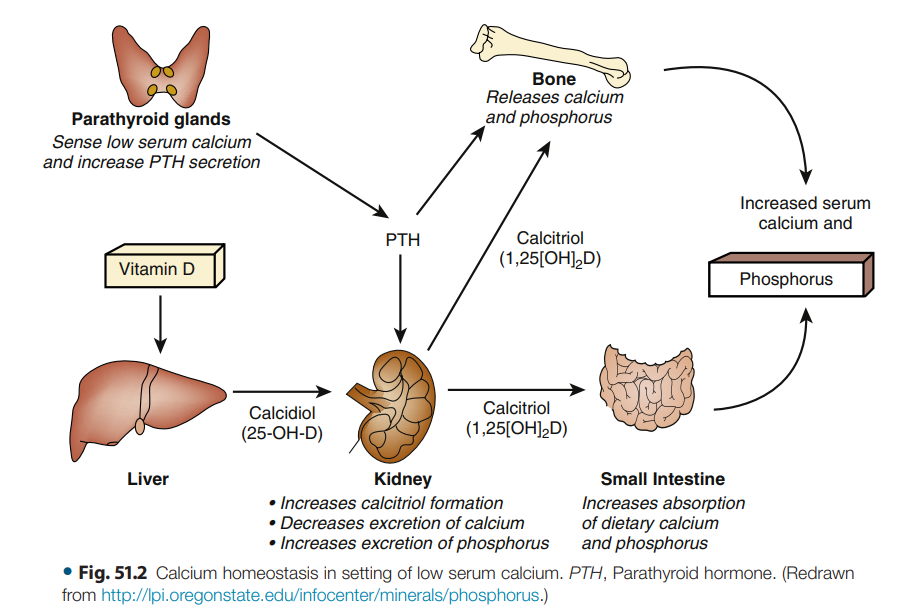
Figura 51.2.
LAS GLÁNDULAS PARATIROIDES Y LA HORMONA PARATIROIDES
Las cuatro glándulas paratiroides derivan de las
bolsas branquiales tercera y cuarta y residen adyacentes a la glándula tiroides
en el cuello. Son muy pequeñas, cada una pesa sólo alrededor de 40 g. La célula
epitelial predominante en las glándulas paratiroides se denomina célula
principal, que tiene un citoplasma claro y es distinta de la célula oxífila más
grande, que tiene un citoplasma granular eosinofílico. Ambos tipos de células
contienen PTH.
Las células paratiroideas “perciben” el nivel de calcio
ionizado por medio de los CaSR que se expresan en la superficie de las células.
La relación entre la concentración de calcio ionizado extracelular y la PTH es
una curva sigmoidea pronunciada en la que pequeños cambios en el calcio
ionizado producen cambios marcados en la PTH (fig. 51.3).

Figura 51.3.
Los cambios en el calcio sérico regulan tanto la
secreción de PTH preformada como la síntesis de novo de PTH al nivel de la
transcripción génica. La vitamina D también juega un papel en la regulación del
gen de la PTH, ya que los niveles altos de calcitriol (1,25[OH]2) inhiben la
transcripción del gen de la PTH. Esto permite utilizar calcitriol o análogos de
la vitamina D en el tratamiento del hiperparatiroidismo secundario (HPT) en
pacientes con insuficiencia renal.
HIPERCALCEMIA
El diagnóstico diferencial de la hipercalcemia se
muestra en el cuadro 51.1. La primera pregunta que debe hacerse en cualquier
caso de hipercalcemia es: ¿cuál es el nivel de PTH? Si la PTH es francamente
alta o incluso inapropiadamente normal en el marco de la hipercalcemia, el
diagnóstico es hiperparatiroidismo primario (HPPT).

Recuadro 51.1. Diagnóstico Diferencial de la Hipercalcemia.
Hay algunos otros diagnósticos que deben ser
considerados. Estos incluyen el HPT terciario (por insuficiencia renal crónica)
y el uso de litio o tiazidas, posibilidades que se eliminan fácilmente. Una
cuarta posibilidad es la condición autosómica dominante muy rara conocida como
hipercalcemia benigna familiar o hipercalcemia hipocalciúrica familiar (FHH).
Las personas con FHH tienen mutaciones inactivadoras del CaSR, lo que hace que
el receptor sea menos sensible o resistente a la concentración sérica ambiental
de calcio tanto en las paratiroides como en los riñones. En otras palabras,
estos individuos requieren niveles más altos de calcio sérico para mantener la
homeostasis normal del calcio. El sello distintivo de FHH es una excreción
urinaria de calcio inapropiadamente baja que se puede calcular fácilmente
usando una fórmula simple:
Fórmula:

La orina CaCl/CrCl <0.01 es consistente con FHH.
Estos pacientes no deben ser enviados para cirugía de paratiroides. Se pueden
realizar pruebas genéticas para mutaciones del CaSR si el diagnóstico es incierto.
HIPERPARATIROIDISMO PRIMARIO
El hiperparatiroidismo primario (HPP), representa del 80% al 90% de la hipercalcemia
en individuos asintomáticos y es, con mucho, la causa más común de
hipercalcemia en pacientes ambulatorios sanos. Es el tercer trastorno endocrino
más común en los Estados Unidos y ocurre en 1 de cada 1000 personas. Las
mujeres comprenden el 75% de los pacientes con HPP, y la edad promedio al
momento del diagnóstico es de 55 años. La gran mayoría (80%) de los casos de HPP
son causados por adenomas solitarios. Alrededor del 15% de los pacientes
tendrán hiperplasia de cuatro glándulas y del 2% al 4% tendrán adenomas
múltiples. La hiperplasia paratiroidea se encuentra con mayor frecuencia en
tres síndromes hereditarios autosómicos dominantes: neoplasia endocrina
múltiple (NEM) 1, NEM 2A y HPT familiar aislado. El carcinoma de paratiroides
representa <0,5% de los casos. La detección genética de MEN1 debe realizarse
en pacientes muy jóvenes con HPP (menores de 30 a 35 años de edad), aquellos
con antecedentes personales o familiares positivos de otros tumores endocrinos
(paratiroides, páncreas, hipófisis) o aquellos con adenomas paratiroideos
atípicos o múltiples. a cualquier edad.
Los signos y síntomas clásicos del HPP se conocen como
“piedras, huesos, gemidos abdominales y gemidos psíquicos” (por su traducción
en inglés: “stones, bones, abdominal groans, and psychic moans). Estos se
enumeran en el Cuadro 51.2.

Recuadro 51.2. Signos y Síntomas del Hiperparatiroidismo Primario.
Antes de la introducción del cribado químico
multifásico, la mayoría de los pacientes con HPP presentaban manifestaciones
renales (cálculos, nefrocalcinosis, insuficiencia renal) y/o la clásica
enfermedad ósea, osteítis fibrosa quística. Ahora, hasta el 85 % de las
personas con HPP son asintomáticas. Los cálculos renales ocurren en menos del
15% de los pacientes con HPP, mientras que el trastorno óseo más común, la
osteoporosis, afecta principalmente los sitios esqueléticos ricos en hueso
cortical (como el tercio distal del radio). Otras manifestaciones del HPP leve
incluyen dispepsia, náuseas y estreñimiento ("gemidos abdominales"),
así como fatiga, letargo, depresión y dificultad para concentrarse
("gemidos psíquicos"). También pueden ocurrir mialgias, debilidad
muscular, condrocalcinosis, poliuria/polidipsia y nicturia. Las manifestaciones
no clásicas del HPP, como la disfunción cardiovascular y neurológica, están
bajo investigación activa en este momento.
Los hallazgos de laboratorio en HPP típicamente
muestran calcio sérico elevado (corregido para albúmina sérica) con una PTH
intacta en suero simultáneamente elevada. Sin embargo, muchos pacientes con HPP
leve tienen niveles séricos de calcio que fluctúan dentro y fuera del rango
normal. Del mismo modo, hasta el 50% de los niveles séricos de PTH pueden estar
en el nivel medio o superior del rango normal, aunque estos niveles siguen
siendo inapropiados en el contexto de la hipercalcemia. Incluso los niveles
normales bajos de PTH pueden estar asociados con adenomas paratiroideos
secretores de PTH. Los niveles séricos de fósforo tienden a estar por debajo de
3,5 mg/dl debido al efecto fosfatúrico de la PTH en los túbulos renales. Tenga
en cuenta que la deficiencia concomitante de vitamina D es muy común en
pacientes con HPP y, en algunos casos, puede enmascarar la hipercalcemia. Las
pautas recientes recomiendan verificar los niveles de 25-hidroxivitamina D en
todos los pacientes con HPP y corregir cualquier deficiencia para mantener los
niveles por encima de 20 ng/mL.
En los últimos años, se ha reconocido una nueva
categoría de HPT, conocida como HPT normocalcémico (HPTN). Los pacientes con PHPTN
tienen niveles persistentemente normales de calcio sérico total e ionizado,
pero niveles elevados de PTH intacta. El diagnóstico diferencial incluye
deficiencia de vitamina D, malabsorción (p. ej., enfermedad celíaca oculta),
enfermedad renal crónica, hipercalciuria y el uso de ciertos fármacos. Una vez
que se han descartado todos estos, se confirma el diagnóstico de HPPN. La
mayoría de estos pacientes se descubren durante un estudio de osteoporosis,
mientras que algunos presentan cálculos renales o fracturas. Alrededor del 20 %
se vuelven hipercalcémicos con el tiempo, y entre el 20 % y el 30 % necesitarán
cirugía paratiroidea.
La paratiroidectomía sigue siendo el tratamiento
definitivo para HPP. Para las personas con HPP leve y asintomático, un taller
internacional de expertos de 2013 desarrolló pautas para la intervención quirúrgica
(Cuadro 51.3).

Recuadro 51.3
Los pacientes que no cumplen ninguno de estos
criterios (hasta el 50% en algunas series) pueden ser monitoreados con calcio
sérico anual, creatinina sérica, tasa de filtración glomerular estimada (TFG) y
densidad mineral ósea (DMO) cada 1 a 2 años ( Recuadro 51.4).

Recuadro 51.4
Los estudios longitudinales de pacientes con HPP
asintomático muestran una notable estabilidad bioquímica durante 10 a 15 años,
aunque hasta el 25 % finalmente requiere cirugía. Después de la curación
quirúrgica del HPP, hay mejoras dramáticas en la DMO y una reducción del 90% al
95% en la formación de cálculos renales en aquellos con nefrolitiasis previa.
Ensayos clínicos recientes que aleatorizaron sujetos con HPP para
paratiroidectomía u observación han encontrado mejoras en la densidad ósea en
los grupos quirúrgicos pero efectos variables en la calidad de vida y los
síntomas.
Para los pacientes no quirúrgicos, el tratamiento
médico incluye una ingesta moderada de calcio de 1000 mg/d (pero más baja en
aquellos con calcitriol alto o niveles altos de calcio en orina), corrección de
la deficiencia de vitamina D, buena hidratación y, en casos seleccionados,
terapia antirresortiva con bisfosfonatos u otros agentes antirresortivos. El
cinacalcet, es un calcimimético aprobado por la Administración de Drogas y
Alimentos de los EE. UU. (FDA) para el HPT primario y secundario y el cáncer de
paratiroides, pero en ensayos recientes ha controlado con éxito la hipercalcemia
en pacientes con HPP durante un máximo de 3 a 5 años. Desafortunadamente,
cinacalcet no mejoró la densidad ósea en pacientes con HPP a pesar de que el
calcio sérico se normalizó y los niveles de PTH se redujeron.
HIPERCALCEMIA NO MEDIADA POR HORMONA PARATIROIDEA
Las etiologías de la hipercalcemia no mediada por PTH
se pueden dividir en tres categorías amplias: de absorción, de reabsorción o
mixta. La hipercalcemia por absorción, por exceso de calcio, se caracteriza por
una mayor absorción de calcio en el intestino. El mejor ejemplo de esto es la
ingestión excesiva de carbonato de calcio que conduce al síndrome lácteo-alcalino.
Los trastornos mixtos con hipercalcemia tanto de absorción como de reabsorción
incluyen hipercalcemia mediada por vitamina D, como intoxicación por vitamina D
exógena o producción excesiva de 1,25dihidroxivitamina D a partir de macrófagos
activados en enfermedades granulomatosas (p. ej., sarcoidosis) o ciertos
linfomas. La hipercalcemia de reabsorción ocurre siempre que la reabsorción
ósea osteoclástica excesiva es el mecanismo principal que subyace a la
hipercalcemia. Aunque varios trastornos benignos pueden estar asociados con
hipercalcemia de reabsorción no mediada por PTH (incluidos hipertiroidismo,
inmovilización, enfermedad de Paget, intoxicación por vitamina A), la mayoría
de los casos son causados por tumores malignos. Los mecanismos de la
hipercalcemia maligna incluyen la liberación local de citocinas activadoras de
osteoclastos en el hueso (mieloma múltiple), la destrucción osteolítica del
hueso por metástasis (cáncer de mama) o la reabsorción esquelética mediada por
el péptido relacionado con la hormona paratiroidea (PTHrP) de tumores distantes
(carcinomas de células renales y escamosas más comúnmente).
Entre los pacientes hospitalizados con hipercalcemia sintomática,
el 45 % tiene neoplasias malignas, el 25 % tiene HPP y el 10 % tiene insuficiencia
renal. Recuerde que el HPP y las neoplasias malignas pueden coexistir en el mismo
paciente. Además, hay varias causas raras de hipercalcemia reportadas en la literatura.
Los síntomas de la hipercalcemia dependen de la
gravedad de la elevación del calcio, así como de la rapidez de la elevación. En
general, los aumentos rápidos de calcio causan más síntomas que los aumentos
más lentos y graduales. Los pacientes con hipercalcemia no mediada por PTH
tienden a tener síntomas más graves que afectan el sistema nervioso central
(letargo, psicosis, estupor y coma), cardiovascular (bradicardia, asistolia y
acortamiento del intervalo QT) y gastrointestinal (anorexia, náuseas, vómitos y
estreñimiento), pero el HPP acelerado también puede causar sintomatología
grave, particularmente en un contexto de insuficiencia renal. Cuando los
niveles de calcio corregidos en suero alcanzan los 14 mg/dl o más, los
pacientes suelen presentar bastantes síntomas.
El estudio del paciente sintomático con hipercalcemia
requiere una anamnesis y un examen físico muy cuidadosos, seguidos de pruebas
de laboratorio juiciosas (tabla 51.1).

Tabla 51.1 Estudio de pacientes con Hipercalcemia no mediada por PTH.
Las pruebas de laboratorio útiles incluyen biometría
hemática completa de rutina, fósforo, magnesio, PTH intacta, 25-hidroxivitamina
D (la prueba clave en la intoxicación por vitamina D), 1,25-dihidroxivitamina D
(la prueba clave en enfermedades granulomatosas y algunos linfomas), y
electroforesis de proteínas séricas y
electroforesis de proteínas en orina (electroforesis de proteínas en orina;
mieloma múltiple). La PTHrP sérica rara vez se necesita para hacer el
diagnóstico de hipercalcemia humoral de malignidad, pero puede ser confirmatoria
en los casos en que el tumor primario es esquivo. Se debe solicitar vitamina A
y cortisol sérico si está clínicamente indicado. Otras pruebas útiles pueden
ser radiografías de tórax, gammagrafía ósea, mamografía, tomografías
computarizadas de tórax/abdomen/pelvis y biopsia de ganglios linfáticos o
tejido.
El manejo de la hipercalcemia sintomática y severa,
independientemente de la causa, comienza con una hidratación vigorosa y la
restauración de la tasa de filtración glomerular a la normalidad, si es posible
(Cuadro 51.5).

Recuadro 51.5. Manejo de la Hipercalcemia Aguda.
Esto mejora la eliminación renal de calcio y reduce
sustancialmente el calcio sérico. Los diuréticos del asa deben usarse para
aumentar la excreción renal de calcio solo después de que se haya restablecido
la euvolemia si hay evidencia de insuficiencia cardíaca o sobrecarga de volumen.
En el síndrome de lácteo alcalina, la hidratación adecuada y el cese de la
fuente de calcio revierten por completo la hipercalcemia. Sin embargo, en la
mayoría de las otras condiciones, la terapia antirresortiva es necesaria para
lograr y mantener la normocalcemia. Los bisfosfonatos intravenosos reducen
rápidamente la hipercalcemia mixta y de reabsorción en el paciente bien
hidratado al provocar la apoptosis de los osteoclastos activados. Tanto el
pamidronato como el ácido zoledrónico pueden reducir el calcio sérico al rango
normal en unos pocos días. Los efectos secundarios adversos de los
bisfosfonatos intravenosos incluyen reacciones de fase aguda, insuficiencia
renal e hipocalcemia. La terapia frecuente con bisfosfonatos intravenosos en
dosis altas para enfermedades malignas puede provocar osteonecrosis de la
mandíbula. Una opción más nueva es denosumab, el anticuerpo monoclonal humano
que se dirige al ligando RANK, una citoquina responsable de la resorción ósea
osteoclástica. Cuando se administra a una dosis de 120 mg por vía subcutánea
cada 4 semanas, el denosumab está aprobado por la FDA para la prevención y el
tratamiento de eventos relacionados con el esqueleto en pacientes con tumores
sólidos metastásicos. Ofrece ciertas ventajas sobre los bisfosfonatos
intravenosos, incluida una excelente tolerabilidad, una administración más
fácil y ausencia de toxicidad renal. Se puede producir hipocalcemia profunda
cuando se administra denosumab a pacientes con insuficiencia renal grave. En
raras ocasiones, se ha informado osteonecrosis de la mandíbula con el fármaco.
Finalmente, en pacientes con hipercalcemia por mieloma múltiple, linfoma,
sarcoidosis o intoxicación por vitamina A o D, los glucocorticoides son
tratamientos extremadamente efectivos. El éxito en el tratamiento de la
hipercalcemia no mediada por PTH depende en última instancia del tratamiento
del trastorno subyacente (Stewart, 2005).
HIPOCALCEMIA
Los trastornos hipocalcémicos pueden deberse a varias
anomalías del sistema regulador del calcio: deficiencia de PTH, respuesta
anormal a la PTH, trastornos de la vitamina D o formación de complejos o
depósito de calcio (cuadro 51.6).


Recuadro 51.6. Diagnóstico Diferencial de la Hipocalcemia.
Los raros trastornos hereditarios de la resistencia a
la vitamina D (raquitismo dependiente de vitamina D y resistente a la vitamina
D) no se discutirán aquí. Tenga en cuenta que la hipoalbuminemia da como
resultado un calcio sérico total bajo debido a una reducción en la fracción de
calcio unida a proteínas; sin embargo, el calcio ionizado permanece normal.
La hipocalcemia puede presentarse dramáticamente con
síntomas de entumecimiento perioral, parestesias, espasmo carpopedal,
convulsiones o tetania, o puede ser relativamente asintomática. La mayoría de
los síntomas son causados por un aumento de la excitabilidad neuromuscular. La
manifestación muscular clásica de la hipocalcemia es el espasmo carpopedal, una
contracción muscular involuntaria dolorosa de las manos en la que hay aducción
del pulgar, flexión de las articulaciones metacarpofalángicas, extensión de las
articulaciones interfalángicas y flexión de las muñecas (fig. 51.4).

Fig. 51.4 Espasmo del carpo en
hipocalcemia.
La tetania también puede presentarse como
laringoespasmo, que puede ser fatal. La tetania latente puede descubrirse
mediante la prueba del signo de Chvostek (golpes en el nervio facial para
producir la contracción de los músculos faciales ipsilaterales) y el signo de
Trousseau (inflar un manguito de presión arterial a 20 mm Hg por encima de la
presión sistólica para provocar espasmo carpiano ipsolateral). Tenga en cuenta
que el 25% de las personas normales pueden tener un signo de Chvostek leve.
Además de la tetania, otras manifestaciones graves de hipocalcemia pueden
incluir convulsiones y prolongación del intervalo QT en las pruebas de
electrocardiograma (debido al retraso en la repolarización), lo que provoca
arritmias graves e insuficiencia cardíaca congestiva.
HIPOPARATIROIDISMO
Los síndromes de hipocalcemia asociados con niveles
bajos de PTH incluyen hipoparatiroidismo posquirúrgico, “síndrome del hueso
hambriento” después de cirugía paratiroidea, hipomagnesemia, enfermedad
crítica, destrucción autoinmune o infiltrativa de las glándulas paratiroides,
así como trastornos hereditarios que incluyen hipocalcemia autosómica
dominante. En este trastorno, el otro lado de FHH, las mutaciones activadoras
de CaSR dan como resultado una mayor sensibilidad al calcio y un punto de
ajuste más bajo para Ca y PTH.
El hipoparatiroidismo quirúrgico es la causa más común
de deficiencia de PTH. Es el resultado de la extirpación o destrucción de las
glándulas paratiroides durante la cirugía por cáncer de cabeza o cuello,
tiroidectomía total o paratiroidectomía. Los estudios de laboratorio muestran
hipocalcemia, hiperfosfatemia y PTH indetectable o inapropiadamente baja. Otras
causas de hipoparatiroidismo como la destrucción autoinmune de las glándulas
paratiroides y los trastornos hereditarios (hipoparatiroidismo familiar,
síndrome de DiGeorge) son mucho menos comunes.
La destrucción infiltrativa de las paratiroides por
hierro (talasemia), cobre (enfermedad de Wilson), enfermedad metastásica o
infecciones también es inusual.
Una causa importante y reversible de
hipoparatiroidismo es la deficiencia grave y crónica de magnesio que paraliza
la secreción de PTH de las vesículas en las glándulas paratiroides, al tiempo
que mitiga las acciones periféricas de la PTH. La hipocalcemia responde
rápidamente a la administración de magnesio.
PSEUDOHIPOPARATIROIDISMO
Esta categoría incluye dos trastornos hereditarios
raros de resistencia de órganos diana a la PTH, denominados
pseudohipoparatiroidismo (PHP) 1A y PHP 1B. Desde el punto de vista bioquímico,
los síndromes se presentan exactamente como el hipoparatiroidismo con
hipocalcemia e hiperfosfatemia, pero la PTH está elevada en lugar de ser
indetectable. PHP 1A está asociado con un fenotipo clásico conocido como
osteodistrofia hereditaria de Albright en la que los pacientes son bajos,
tienen caras redondas, cuellos cortos, retraso mental y acortamiento del cuarto
y/o quinto metacarpianos. Los pacientes con PHP 1B no tienen fenotipo somático
y, por lo demás, parecen normales. Las mutaciones genéticas y la herencia de
ambos síndromes ahora se han dilucidado por completo.
DEFICIENCIA DE VITAMINA D
Los pacientes con hipocalcemia causada por deficiencia
de vitamina D tienen niveles elevados de PTH (el llamado HPT secundario). Las
causas más comunes de deficiencia de vitamina D se muestran en el Cuadro 51.7.

Recuadro 51.7. Causas de Deficiencia de Vitamina D.
Tenga en cuenta que la hipocalcemia rara vez ocurre
por la depleción de 25-hidroxivitamina D sola, incluso cuando los niveles son
indetectables, debido a la capacidad de los niveles altos de PTH para movilizar
el calcio del hueso.
ENFERMEDAD RENAL CRÓNICA
Una de las causas más importantes del HPT secundario
es la enfermedad renal crónica (ERC). Los pacientes en diálisis a menudo tienen
niveles extremadamente elevados de PTH y fragmentos de PTH (no todos son
bioactivos) causados por la disminución de los niveles de
1,25-dihidroxivitamina D y aumentos en el fosfato sérico asociado con
insuficiencia renal progresiva. El metabolismo mineral desordenado asociado con
la ERC puede resultar en osteodistrofia renal, calcificaciones vasculares y de
tejidos blandos, enfermedad cardiovascular y alta mortalidad cardiovascular. En
el pasado, los únicos tratamientos disponibles para el HPT secundario
resultante de la ERC eran grandes dosis de calcio para que sirvieran como
aglutinantes de fosfato, así como esteroles de vitamina D para reducir la PTH.
Estas intervenciones a menudo agravaron el metabolismo mineral anormal al
aumentar el producto calcio × fosfato y empeorar la calcificación vascular y
ectópica. El calcimimético cinacalcet ha ofrecido una atractiva alternativa al
tratamiento tradicional del HPT secundario en la ERC. Al imitar el calcio en el
CaSR, cinacalcet reduce la PTH sin aumentar el calcio sérico, el fosfato o el
producto calcio x fosfato. Ha mejorado mucho la homeostasis del fosfato de calcio
en pacientes en diálisis.
MISCELÁNEAS
Varios otros trastornos pueden provocar hipocalcemia,
incluida la hiperfosfatemia por rabdomiolisis o síndrome de lisis tumoral,
malabsorción de calcio por enfermedad celíaca u otros estados de malabsorción, transfusiones
con sangre citratada, metástasis esqueléticas osteoblásticas diseminadas y
pancreatitis aguda, entre otros. La alcalosis respiratoria aguda por
hiperventilación puede causar hipocalcemia reversible sintomática causada por
un cambio de calcio ionizado en albúmina dentro del entorno alcalótico.
TRATAMIENTO
El tratamiento de la hipocalcemia depende de la causa,
la gravedad y el grado de sintomatología, pero generalmente incluye suplementos
de calcio y vitamina D. La hipocalcemia aguda asociada con tetania o tetania
incipiente es una emergencia médica y requiere la administración intravenosa
inmediata de calcio (Cuadro 51.8).

Recuadro 51.8. Manejo de la Hipocalcemia.
Se pueden administrar de dos a tres ampollas de
gluconato de calcio (90 mg de calcio elemental por ampolla de 10 ml) por vía
intravenosa durante varios minutos, seguidas de una infusión de 10 ampollas en
1 L de líquidos intravenosos durante 24 horas. Simultáneamente, el paciente
debe iniciar calcitriol (vitamina D activada) y calcio oral. Para la
hipocalcemia crónica causada por hipoparatiroidismo, el objetivo es mantener el
calcio sérico en el rango de 8,5 a 9 mg/dL, lo suficientemente alto para
prevenir los síntomas pero lo suficientemente bajo para evitar la
hipercalciuria. Es deseable el control periódico de la excreción de calcio en
orina de 24 horas para detectar hipercalciuria. El tratamiento con diuréticos
tiazídicos puede ayudar a reducir la excreción urinaria de calcio. El objetivo
es mantener los niveles de calcio en orina de 24 horas por debajo de 4 mg/kg/día.
Deben administrarse al menos 1,5 a 3 g de calcio elemental oral diariamente en
dosis divididas junto con vitamina D activada (calcitriol). Se pueden usar
grandes dosis de ergocalciferol (vitamina D2) o colecalciferol (vitamina D3) en
lugar de calcitriol, pero pueden acumularse y causar intoxicación por vitamina
D.
En 2015, la FDA aprobó la PTH humana recombinante
(1-84) para el tratamiento de pacientes con hipocalcemia resultante de
hipoparatiroidismo permanente. El fármaco se administra como inyección
subcutánea todos los días y permite a los pacientes lograr un mejor control de
la calcemia con dosis más bajas de calcio y vitamina D. Sin embargo, la rhPTH
(1-84) es bastante costosa y viene con una advertencia sobre osteosarcoma en
ratas. Es útil para pacientes con hipoparatiroidismo que no pueden lograr un
control razonable de su síndrome de deficiencia con terapias sin PTH.
Fuente: "The
Brigham Intensive Review of
Internal Medicine". (2022)
Ajay K. Singh, MBBS, FRCP, MBA
Joseph Loscalzo, MD, PhD