Paciente femenino de 15 años de edad, natural de la
serranía peruana, estatus socioeconómico bajo, hacinamiento en vivienda, aves
de corral en casa; no antecedentes patologicos de importancia, aparentemente
sana. Acude el día de hoy al centro de salud refiriendo textualmente lo
siguiente "desde hace 1 año “heridas” en pierna y pie izquierdo que
inician como ampollas, las cuales se vuelven duras en los 2 a 3 días siguientes
con bastante escozor, el cual perdura hasta el momento"; refiere además
que en el último mes han incrementado en cantidad y tamaño. Tratamiento
recibido: clotrimazol tópico por 3 meses,
(aparentemente tratamiento regular), posteriormente terbinafina crema
por 4 meses tratamiento regular; hace 10 dias dicloxacilina 500mg por 7 dias
más betametasona 0.05% tópico por 10 días; sin notar mejoría con ninguno de
ellos. Examen físico: se palpan lesiones nodulares, no dolorosas de aprox 1 a
1.5 cm de diámetro algunas redondeadas, otras ovales, conglomeradas, pruriginosas
al contacto con el guante en 1/3 distal de pierna izquierda y dorso de pie
izquierdo. No exámenes auxiliares.
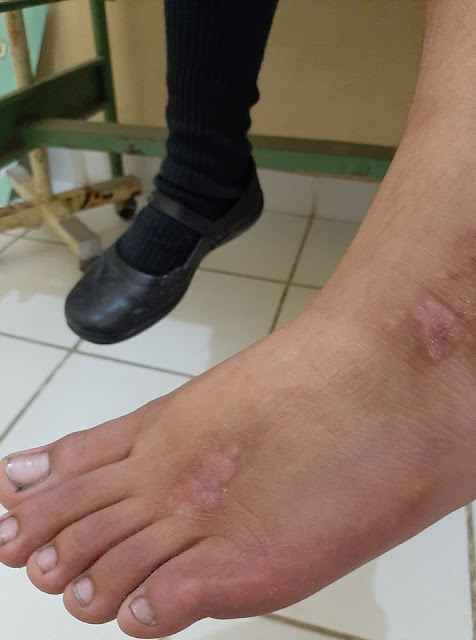
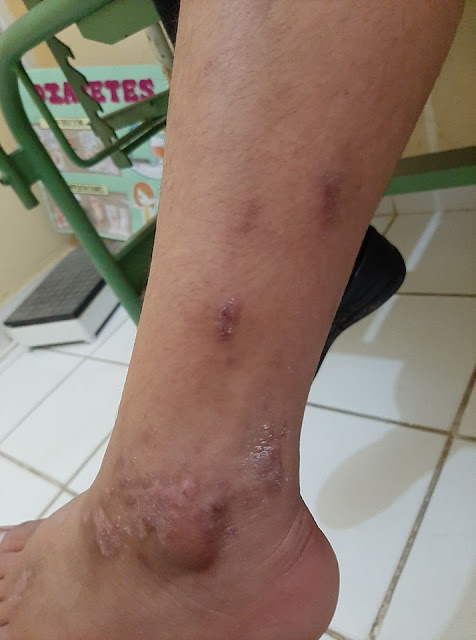

El cuadro es típico de liquen simple crónico. El
hecho de que aparezcan inicialmente ampollas podría indicar picaduras de
insectos como desencadenante primario. Le anexo algunas fotos que tomé en
consulta. Gracias de antemano.
Gentileza:
Dr. Christian
Henry Juárez Carreño
Piura, Perú.
LIQUEN SIMPLE CRÓNICO
El liquen simple crónico (LSC, por sus siglas en
inglés) es un engrosamiento de la piel con descamación variable que surge como
consecuencia de rascarse o frotarse repetidamente. El liquen simple crónico no
es un proceso primario. Más bien, una persona siente prurito en un área
específica de la piel (con o sin patología subyacente) y causa un trauma
mecánico hasta el punto de liquenificación.
Una variante propuesta de liquen simple crónico es
la amiloidosis liquen . Liquen amiloidosis se describe como liquen simple
crónico en el que los queratinocitos se han necrosado y formado amiloide
derivado de queratinocitos en la dermis. El insulto inicial es el prurito con
la formación de amiloide resultante, en lugar a la inversa. [ 1 , 2 ]
FISIOPATOLOGÍA
El liquen simple crónico se encuentra en la piel en
regiones accesibles al rascado. El prurito provoca un rascado que a su vez produce lesiones clínicas, pero se
desconoce la fisiopatología subyacente. Algunos tipos de piel son más propensas
a la liquenificación, como la piel que tiende hacia condiciones eccematosas (
dermatitis atópica, diátesis atópica). Es probable que exista una relación
entre el tejido neural central y periférico y los productos de células
inflamatorias en la percepción de la picazón y los cambios resultantes en el
liquen simple crónico. Las tensiones emocionales, como en pacientes con
ansiedad, depresión o trastorno obsesivo compulsivo, pueden desempeñar un papel
clave en la inducción de una sensación pruriginosa, lo que puede provocar un
rascado que puede perpetuarse. [ 3 , 4 , 5 , 6] La posible interferencia entre
las lesiones primarias, factores psíquicos, y la intensidad del prurito
influyen de forma aditiva la extensión y gravedad de liquen simple crónico.
Un pequeño estudio en liquen simple crónico y el uso
de pelo desecado que contiene P-fenilendiamina (PPD) mostró una mejoría
clínicamente relevante en los síntomas después de la interrupción de la
exposición a PPD, [ 7 ] proporcionando así una base para el papel de la
sensibilización y la dermatitis de contacto en La etiología del liquen simple
crónico.
ETIOLOGÍA
La dermatitis atópica genera una mayor probabilidad
de desarrollar liquen simple crónico.
Las picaduras de insectos, cicatrices (p. Ej.,
Traumáticas, postherpéticas / zoster [ 8 ] ), acné queloide nucae, [ 9 ]
xerosis, insuficiencia venosa y eccema asteatósico son factores comunes.
La neurodermatitis es un término que históricamente
se ha utilizado indistintamente con el liquen simple crónico, dado que los
factores psicológicos parecen desempeñar un papel en el desarrollo o la
exacerbación de la afección. [ 10 , 11 , 12 ] Un estudio de 2014 demostró un
aumento de la prevalencia de liquen simple crónico en pacientes con ansiedad
subyacente y trastorno obsesivo compulsivo en comparación con los controles
pareados por edad y sexo. [ 5 ] Un estudio de 2015 mostró que los pacientes con
liquen simple crónico tienen mayores tasas de depresión clínica en comparación
con los pacientes sin la afección. [ 6 ]
El litio se ha relacionado con el liquen simple
crónico en un caso reportado. El liquen simple crónico dependía de la
administración de litio, como lo demuestra la observación de que el liquen
simple crónico remitía cuando se descontinuó la medicación y recurrió cuando se
reinició. [ 13 ]
Un pequeño estudio que analizó el liquen simple
crónico y el uso de pelo desecado con PPD mostró una mejoría clínicamente
relevante en los síntomas después de la interrupción de la exposición al PPD,
proporcionando así una base para el papel de la sensibilización y la dermatitis
de contacto en la etiología del liquen simple crónico. [ 7 ] . Varios de los pacientes
que desaparecieron después de evitar la parafenilendiamina tenían dermatitis
generalizada, no localizada.
Algunos se reservan el diagnóstico de liquen simple
para pacientes que no tienen un trastorno cutáneo predisponente conocido. El
término liquenificación secundaria se ha utilizado si la erupción se inicia por
una dermatosis primaria.
EPIDEMIOLOGÍA
FRECUENCIA
La frecuencia exacta en la población general es
desconocida. En un estudio, el 12% de los pacientes mayores con piel
pruriginosa tenían liquen simple crónico.
RAZA
No se ha visto diferencias en la frecuencia entre
razas, aunque los autores anteriores afirmaron que el liquen simple crónico era
más común en asiáticos y afroamericanos. La aparición de lesiones en la piel
más oscura a veces muestra prominencia folicular. Las alteraciones pigmentarias
secundarias también son más graves en personas con piel más oscura.
SEXO
El liquen simple crónico se observa con mayor
frecuencia en mujeres que en hombres. El liquen nucal es una forma de liquen
simple que ocurre en el cuello posterior medio y se observa casi exclusivamente
en mujeres.
AÑOS
El liquen simple crónico se presenta principalmente
en la edad adulta media a tardía, con la mayor prevalencia en personas de 30 a
50 años.
PRONÓSTICO
Las lesiones pueden desaparecer por completo. El
prurito puede resolverse, pero quedan algunas cicatrices leves y cambios
pigmentarios después del tratamiento exitoso. La recaída es más probable en
períodos de estrés psíquico o si la piel afectada anteriormente está estresada
por los extremos de calor o humedad o por irritantes o alérgenos de la piel. En
pacientes que no cumplen con el régimen de tratamiento y la interrupción del
rascado, las lesiones no mejorarán.
No se produce mortalidad como resultado del liquen
simple crónico. En general, el prurito del liquen simple crónico es de leve a
moderado, pero pueden producirse paroxismos que se alivian con roces y rascarse
de moderados a severos. El prurito generalmente se describe como mucho peor
durante los períodos de inactividad, generalmente al acostarse y durante la
noche. [ 15 ] El tacto y el estrés emocional también pueden provocar prurito,
que se alivia con roces y rascarse de moderados a severos.
Las lesiones causan poca morbilidad directa; sin
embargo, ocasionalmente los pacientes informan una disminución o interrupción
del sueño, lo que afecta el funcionamiento motor y mental.
El liquen simple crónico puede infectarse
secundariamente después de la excoriación.
El liquen simple crónico a menudo es lo suficientemente
visible como para hacer que los pacientes busquen tratamiento.
EDUCACIÓN DEL PACIENTE
Dirija a los pacientes a que dejen de rascarse. El
liquen simple crónico empeora o mejora dependiendo de la capacidad del paciente
para dejar de rascarse. Discutir formas individuales de cambiar el rascado
habitual es útil.
CLÍNICA
Los pacientes con liquen simple crónico generalmente
describen placas pruriginosas estables en una o más áreas; sin embargo, el
engrosamiento de la piel se produce en cualquier lugar que el paciente pueda
alcanzar, incluidos los siguientes:
- Cuero cabelludo [ 16 ]
- Nuca
- Superficie de extensión de antebrazos y codos
- Vulva y escroto [ 6 , 17 , 18 , 19 , 20 ]
- Región medial y media superior de muslos, rodillas,
piernas y tobillos
- El eritema se nota más en las lesiones tempranas.
- El prurito se describe como peor cuando los pacientes
están quietos y mucho menos o inexistente cuando los pacientes están activos.
- El prurito suele ser intermitente; El rascado
resultante proporciona un alivio temporal.
- Los pacientes pueden tener un historial médico
anterior de una afección crónica de la piel o un trauma agudo. Los pacientes
con dermatitis atópica pueden tener liquen simple crónico en áreas de brotes
atópicos anteriores. Los sitios de dermatitis de contacto alérgica o irritante,
picaduras de insectos u otros traumatismos cutáneos menores en ocasiones
muestran prurito y, posteriormente, liquen simple crónico.
EXAMEN FÍSICO
Se observan una o más placas ligeramente
eritematosas, escamosas, bien delimitadas, liquenificadas, firmes y ásperas con
líneas de piel exageradas.
Cada placa del tamaño de una palma puede tener 3
zonas. Una zona periférica de 2 a 3 cm de ancho que apenas está engrosada puede
tener pápulas aisladas. La zona media tiene pápulas prurigo lenticulares y
hemisféricas que pueden estar excoriadas. La zona central tiene el mayor
engrosamiento y alteración pigmentaria.
Los cambios pigmentarios (especialmente la
hiperpigmentación) se observan de forma variable como en cualquier lesión
dermatítica.
El frotado y rascado juega un papel clave en la formación de la
lesión y se visualiza de forma variable por las marcas blancas de arañazos, la erosión
y la ulceración causada por el rascado más profundo.
El liquen simple crónico es uno de los procesos
hiperqueratóticos a partir del cual puede crecer un cuerno cutáneo. [ 21 ]
Los pacientes pueden rascarse las lesiones de novo
cuando se observan. Algunos pacientes pueden comenzar a rascarse mientras
discuten la picazón o describen las lesiones.
Imagen 4: Liquen simple crónico de la mano y la
muñeca dorsales que demuestran un aumento del grosor de la piel y la
acentuación de las marcas de la piel.
Imagen 5: Placa de liquen simple crónico de la
pierna con marcas cutáneas acentuadas y excoriaciones.
Imagen 6: Placas de liquen simple crónico en la
mano.
Imagen 7: Placa de liquen simple crónico que muestra
marcas cutáneas acentuadas
Imagen 8: Área del liquen simple crónico que
originalmente se creía que era dermatitis de contacto crónica. La verdadera
naturaleza se reveló cuando el paciente admitió frotar esta área mientras
miraba televisión
COMPLICACIONES
Los pacientes con liquen simple crónico tienen tasas
más altas de diabetes mellitus, hiperlipidemia, hipertensión, enfermedad
arterial coronaria, enfermedad vascular periférica, enfermedad pulmonar
obstructiva crónica y enfermedad renal crónica. [ 6 ]
Los pacientes con liquen simple crónico tienen más
probabilidades de desarrollar disfunción eréctil que los controles de la misma
edad. Los médicos deben conocer la asociación entre el liquen simple crónico y
la disfunción eréctil y aconsejar u organizar consultas sexuales oportunas. [ 6
]
DIAGNÓSTICO DIFERENCIAL
- Acantosis nigricans
- Acné Keloidalis Nuchae (AKN)
- Dermatitis de contacto alérgica
- Alopecia Mucinosa
- Dermatitis de Berloque
- Linfoma cutáneo de células T
- Dermatitis herpetiforme
- Manifestaciones dermatológicas de enfermedad
gastrointestinal
- Manifestaciones dermatológicas de la enfermedad
hematológica.
- Manifestaciones dermatológicas de la enfermedad
neurológica.
- Manifestaciones dermatológicas de enfermedad renal
- Enfermedad de Paget extramamaria
- Hiperqueratosis del pezón y areola
- Dermatitis de contacto irritante
- Liquen Nitidus
- Liquen plano
- Liquen estriado
- Dermatitis numular (eccema numular)
- Dermatitis atópica pediátrica
- Fitofotodermatitis
- Psoriasis en placas
- Melanosis de Riehl (dermatitis de contacto
pigmentada)
- Dermatitis seborreica
- Dermatitis por estasis
- Tiña Cruris
LABORATORIO
ESTUDIOS DE LABORATORIO
Un nivel elevado de inmunoglobulina E sérica
ocasionalmente respalda el diagnóstico de una diátesis atópica subyacente.
Realice un examen de hidróxido de potasio y cultivos de hongos para excluir la
tiña crural o la candidiasis en pacientes con liquen simple crónico genital.
OTRAS PRUEBAS
Las pruebas de parche ayudan a excluir la dermatitis
de contacto alérgica como una dermatosis primaria subyacente (p. Ej.,
Dermatitis de contacto alérgica al níquel con liquen simple crónico secundario)
o como factor de cronicidad (p. Ej., Dermatitis de contacto alérgica a
corticosteroides tópicos utilizados para tratar el liquen simple crónico).
PROCEDIMIENTOS
Con frecuencia, la biopsia de piel se realiza para
excluir otros trastornos, particularmente la psoriasis o la micosis fungoide
(linfoma cutáneo de células T) en pacientes de edad avanzada. [ 22 ]
HALLAZGOS HISTOLÓGICOS
El examen histológico demuestra hiperqueratosis,
acantosis, espongiosis y parches de paraqueratosis en la epidermis. Se observa
engrosamiento epidérmico de todas las capas, con alargamiento de las crestas
rete y con hiperplasia pseudoepiteliomatosa. La fibrosis dérmica papilar con
rayas verticales de haces de colágeno es característica.
Un hallazgo característico del liquen simple crónico
que se observa en la microscopía electrónica son las fibras de colágeno
frecuentes unidas a la lámina basal y justo por encima de esta.
Imagen 9: Biopsia H y E de liquen simple crónico de
la piel del antebrazo vista con un aumento de 40x. Obsérvese la hiperqueratosis
característica, hipergranulosis, hiperplasia pseudoepiteliomatosa y fibrosis
dérmica papilar.
Imagen 10: Biopsia H y E de liquen simple crónico de
la piel del antebrazo vista con un aumento de 100x. Obsérvese la
hiperqueratosis característica, hipergranulosis, hiperplasia
pseudoepiteliomatosa y fibrosis dérmica papilar.
TRATAMIENTO
El tratamiento está dirigido a reducir el prurito y
la minimización de las lesiones existentes debido a frotar o rascar causa del
liquen simple crónico. Ubicación, morfología de la lesión, y el alcance del
tratamiento de las lesiones influencia. Por ejemplo, una placa psoriasiforme
espesor de liquen simple crónico en una extremidad se trata comúnmente con corticosteroides muy potentes intralesionales
o tópicos, mientras que las lesiones vulvares son tratados más comúnmente con
un corticosteroide tópico suave o un inhibidor de la calcineurina tópico.
lesiones generalizadas son más propensas a requerir tratamiento sistémico o
fototerapia corporal total.
Los esteroides tópicos son el tratamiento actual de
elección debido a que disminuyen la inflamación y picazón mientras
simultáneamente suavizan la hiperqueratosis. [ 23 , 24 , 25 ]En lesiones más
grandes y más activas, un esteroide potencia media se puede usar para tratar la
inflamación aguda. De vez en cuando, la oclusión se utiliza para aumentar la
potencia y mejorar el suministro del agente. La oclusión también proporciona
una barrera física para el rascado. Los esteroides tópicos de potencia media no
se recomiendan para la piel delgada (por ejemplo, la vulva, el escroto, la
axila, la cara). Dirigir a largo plazo la terapia más en el uso diario de corticosteroides
de baja potencia no trofogénicos. Los corticosteroides tópicos de alta potencia pueden ser utilizados para cursos de 3 semanas en las áreas más gruesas
de piel clara.
Inyección intralesional de corticosteroides es útil
para lesiones refractarias. Un informe de 2018 describe el uso de un
dispositivo de inyección neumática transcutánea para lesiones refractarias a 1
mes de tratamiento con un esteroide tópico potente. Triamcinolona inyecta en
las lesiones a intervalos de 2 cm conducido a una mejora significativa. [ 26 ]
Medicamentos contra la ansiedad y la sedación por
vía oral pueden ser considerados en ciertos pacientes. De acuerdo a las
necesidades individuales, el tratamiento puede ser programado durante todo el
día, la hora de acostarse, o ambos. Antihistamínicos como difenhidramina
(Benadryl) y hidroxizina (Atarax) Se utilizan comúnmente. Doxepina (Sinequan) y
clonazepam (Klonopin) pueden ser considerados en los casos apropiados.
Para las lesiones infectadas, un antibiótico tópico
u oral puede ser considerado.
Otros medicamentos tópicos que disminuyen el prurito
incluyen la crema de doxepina [ 27 ] y la crema de capsaicina. [ 28 ]
Un estudio sugiere que la aspirina tópica /
diclorometano es eficaz en pacientes con liquen simple crónico que no han
respondido a los corticosteroides tópicos. [ 29 ]
Para los pacientes que no responden a los
corticosteroides tópicos o aquellos con lesiones en la piel delgada, algunos
informes de casos y pequeños estudios han demostrado la eficacia de los
inmunomoduladores tópicos tacrolimus y pimecrolimus. [ 30 ]
Un tratamiento más investigación para pacientes que
no responden al tratamiento convencional es inyecciones locales de toxina
botulínica. [ 31 , 32 ]
De estimulación nerviosa eléctrica transcutánea
(TENS) ha sido reportado como un posible tratamiento eficaz en un pequeño
ensayo abierto de los casos de liquen simple crónico resistente al tratamiento
con corticosteroides tópicos. [ 33 ]
CONSULTAS
La consulta con un dermatólogo puede ser considerado
para los casos graves que requieren más de los tratamientos tópicos o para
facilitar la prueba del parche.
La consulta con un alergólogo puede considerarse en
individuos con síntomas atópicos multisistémicas.
La consulta con un psiquiatra puede considerarse,
dada la asociación con subyacente trastornos depresivos y de ansiedad.
PREVENCIÓN
Dirija a los pacientes a que dejen de rascarse. El
liquen simple crónico empeora o mejora dependiendo de la capacidad del paciente
para dejar de rascarse. Discutir formas individuales de cambiar el rascado
habitual es útil.
Los extremos de temperatura y / o humedad, estrés
psíquico, y la exposición de las zonas previamente afectadas o predispuesto a
irritantes cutáneos y alérgenos provocan la recaída.
MONITOREO A LARGO PLAZO
Examine periódicamente los pacientes con liquen
simple crónico en una clínica de dermatología ambulatoria para evaluar las
lesiones para los cambios. Realizar el seguimiento de los exámenes con más
frecuencia en pacientes que están siendo tratados con potente clase I
corticosteroides tópicos o agentes orales.
Fuente
Medscape.
REFERENCIAS
Weyers W, Weyers
I, Bonczkowitz M, Díaz-Cascajo C, Schill WB. Liquen
amiloidoso: consecuencia del rascado. J Am Acad Dermatol . 37 de diciembre de
1997 (6): 923-8. [Medline] .
Weyers W. [Liquen amiloidoso - entidad de la
enfermedad o el efecto de rascarse]. Hautarzt . 1995 46 de marzo (3): 165-72.
[Medline] .
Lotti T, Buggiani G, Prignano F. Prurigo nodularis y
liquen simple crónico. Dermatol Ther . 2008 enero-febrero. 21 (1): 42-6.
[Medline] .
Tsintsadze N, Beridze L, Tsintsadze N, Krichun Y,
Tsivadze N, Tsintsadze M. Aspectos psicosomáticos en pacientes con enfermedades
dermatológicas. Georgian
Med News . 2015 junio 70-5. [Medline] .
Liao YH, Lin CC,
Tsai PP, Shen WC, Sung FC, Kao CH. Mayor riesgo de liquen
simple crónico en personas con trastorno de ansiedad: un estudio de cohorte
retrospectivo a nivel nacional basado en la población. Fr. J. Dermatol . 2014 abril 170 (4): 890-4. [Medline]
.
Juan CK, Chen
HJ, Shen JL, Kao CH. Liquen simple crónico asociado con la
disfunción eréctil: un estudio de cohorte retrospectivo de base poblacional.
PLoS One . 2015. 10 (6):
e0128869. [Medline] .
Chey WY, Kim KL, Yoo TY, Lee AY. Dermatitis de
contacto alérgica por tinte capilar y desarrollo de liquen simple crónico.
Dermatitis de contacto . 2004 Jul. 51 (1): 5-8. [Medline] .
Gerritsen MJ, Gruintjes FW, Andreissen MA, van der
Valk PG, van de Kerkhof PC. Liquen simple crónico como complicación del herpes
zoster. Fr. J. Dermatol . 1998 mayo. 138 (5): 921-2. [Medline] .
Burkhart CG, Burkhart CN. El acné keloidalis es
liquen simple crónico con cicatrices queloidales fibróticas. J Am Acad Dermatol
. 1998, 39 de octubre (4 Pt 1): 661. [Medline] .
Woodruff PW, Higgins EM, du Vivier AW, Wessely S.
Enfermedad psiquiátrica en pacientes remitidos a una clínica de dermatología y
psiquiatría. Gen Hosp Psychiatry . 19 de enero de 1997 (1): 29-35. [Medline] .
Gupta MA, Vujcic B, Gupta AK. Síntomas de
disociación y conversión en dermatología. Clin Dermatol . 2017 mayo - 35 de
junio (3): 267-272. [Medline] .
Charifa A, Badri T. Lichen, Simplex Chronicus.
StatPearls [Internet] . 2018 enero. Publicación de StatPearls: Treasure Island,
Florida [Medline] . [Texto completo] .
Shukla S, Mukherjee S. Lichen simplex chronicus
durante el tratamiento con litio. Soy J Psiquiatría . 1984, 141 de julio (7):
909-10. [Medline] .
Ising H, Lange-Asschenfeldt H, Lieber GF, Weinhold
H, Eilts M. [Efectos de la exposición a largo plazo al escape del tráfico de la
calle en el desarrollo de enfermedades de la piel y del tracto respiratorio en
niños]. Schriftenr Ver Wasser Boden Lufthyg . 2003. 81-99. [Medline] .
Boozalis E, Grossberg AL, Püttgen KB, Cohen BA,
Kwatra SG. Picazón en la noche: una revisión sobre la reducción del prurito
nocturno en niños. Pediatr Dermatol . 26 de junio de 2018 [Medline] .
Muylaert BPB, Borges MT, Michalany AO, Scuotto CRC.
Liquen simple crónico en el cuero cabelludo: hallazgos clínicos,
dermatoscópicos e histopatológicos exuberantes. Un Dermatol Bras . 2018 enero-febrero.
93 (1): 108-110. [Medline] .
Ball SB, Wojnarowska F. Dermatosis vulvar: liquen
escleroso, liquen plano y dermatitis vulvar / liquen simple crónico. Semin
Cutan Med Surg . 1998 17 de septiembre (3): 182-8. [Medline] .
Pleimes M, Wiedemeyer K, Hartschuh W. [Liquen simple
crónico de la región anal y sus diagnósticos diferenciales: una serie de
casos]. Hautarzt . 2009 7 de marzo. [Medline] .
Simonetta C, Burns EK, Guo MA. Dermatosis vulvar:
una revisión y actualización. Mo Med . 2015 julio-agosto. 112 (4): 301-7.
[Medline] .
Guerrero A, Venkatesan A. Dermatosis vulvares
inflamatorias. Clin Obstet Gynecol . 58 de septiembre de 2015 (3): 464-75.
[Medline] .
Khaitan BK, Sood A, Singh MK. Liquen simple crónico
con un cuerno cutáneo. Acta Derm Venereol . 1999 mayo. 79 (3): 243. [Medline] .
Thaipisuttikul Y. Enfermedades cutáneas pruriginosas
en ancianos. J Dermatol . 1998 25 de marzo (3): 153-7. [Medline] .
Brunner N, Yawalkar S. Un ensayo doble ciego,
multicéntrico, de grupos paralelos con ungüento de propionato de halobetasol al
0.05% versus ungüento de valerato de diflucortolona al 0.1% en pacientes con
dermatitis atópica crónica grave o liquen simple crónico. J Am Acad Dermatol .
25 de diciembre de 1991 (6 Pt 2): 1160-3. [Medline] .
Datz B, Yawalkar S. Un ensayo doble ciego,
multicéntrico de ungüento de propionato de halobetasol al 0.05% y ungüento de
propionato de clobetasol al 0.05% en el tratamiento de pacientes con dermatitis
atópica crónica localizada o liquen simple crónico. J Am Acad Dermatol . 25 de
diciembre de 1991 (6 Pt 2): 1157-60. [Medline] .
Geraldez MC, Carreon-Gavino M, Hoppe G, Costales A.
Ungüento de valerato de diflucortolona con y sin oclusión en liquen simple
crónico. Int J Dermatol . 28 de noviembre de 1989 (9): 603-4. [Medline] .
Jung HM, Eun SH, Lee JH, Kim GM, Bae JM. Método de
inyección intralesional menos dolorosa y eficaz para el liquen simple crónico.
J Am Acad Dermatol . 79 de diciembre de 2018 (6): e105-e106. [Medline] .
Drake LA, Millikan LE. El efecto antiprurítico de la
crema de doxepina al 5% en pacientes con dermatitis eccematosa. Grupo de
estudio de doxepina. Arch Dermatol . 1995 dic. 131 (12): 1403-8. [Medline] .
Kantor GR, Resnik KS. Tratamiento del liquen simple
crónico con crema de capsaicina tópica. Acta Derm Venereol . 1996 marzo 76 (2): 161. [Medline]
.
Yosipovitch G,
Sugeng MW, Chan YH, Goon A, Ngim S, Goh CL. El efecto de la
aspirina aplicada tópicamente en la neurodermatitis circunscrita localizada. J
Am Acad Dermatol . 45 de diciembre de 2001 (6): 910-3. [Medline] .
Kelekci HK, Uncu HG, Yilmaz B, Ozdemir O, Sut N,
Kelekci S. Pimecrolimus crema al 1% para el prurito en mujeres diabéticas
posmenopáusicas con liquen simple crónico vulvar: una serie de casos
prospectiva no controlada. J Dermatolog Treat . 2008 11 de abril. 1-5.
[Medline] .
Heckmann M, Heyer G, Brunner B, Plewig G. Inyección
de toxina botulínica tipo A en el tratamiento del liquen simple: un estudio
piloto abierto. J Am Acad Dermatol . 2002 Abr. 46 (4): 617-9. [Medline] .
Messikh R, Atallah L, Aubin F, Humbert P. [Toxina
botulínica en enfermedades dermatológicas discapacitantes]. Ann Dermatol
Venereol . 2009 mayo. 136 Supl. 4: S129-36. [Medline] .
Engin B, Tufekci O, Yazici A, Ozdemir M. El efecto
de la estimulación nerviosa eléctrica transcutánea en el tratamiento del liquen
simple: un estudio prospectivo. Clin Exp Dermatol . 2009 abril 34 (3): 324-8.
[Medline] .



























