Las plaquetas sanguíneas, al interactuar con una red compleja de proteínas de coagulación, forman el sistema hemostático, que proporciona la principal defensa del cuerpo contra el exceso de sangrado después de una lesión, cirugía u otros episodios invasivos. Los trastornos del sistema hemostático son una mezcla de enfermedades comunes y raras, hereditarias y adquiridas, leves o potencialmente mortales. Además, aunque el exceso de sangrado es causado por una falla en el sistema hemostático, los pacientes que presentan trombosis pueden tener un defecto en los mecanismos reguladores que normalmente limitan la respuesta hemostática. Ha habido un progreso sustancial tanto en el diagnóstico como en el tratamiento de los trastornos hemostáticos. Además, se han desarrollado fármacos antiplaquetarios y anticoagulantes altamente efectivos para el tratamiento de pacientes con tromboembolismo venoso y arterial, incluidas las enfermedades coronarias y cerebrovasculares.
Este capítulo comienza con un resumen del proceso de
hemostasia normal y revisa las pruebas de laboratorio utilizadas para evaluar
la hemostasia. Luego revisa la fisiopatología, la presentación clínica, el
diagnóstico y el tratamiento de los trastornos hemostáticos más importantes.
Aunque el diagnóstico y el tratamiento dependen en gran medida de las pruebas
de laboratorio, es crucial enfatizar la importancia crítica de la historia
clínica y el examen físico para evaluar a los pacientes con sospecha de un
trastorno hemostático. Una historia cuidadosa proporcionará una evaluación de
la probabilidad de un trastorno y, a veces, es positiva incluso cuando las
pruebas de detección iniciales son normales. Además, la historia puede ayudar a
centrar el estudio en las plaquetas o las proteínas de la coagulación. El
examen físico puede proporcionar pistas importantes sobre la naturaleza del
trastorno hemorrágico y no debe pasarse por alto.
Hemostasia normal
El proceso de hemostasia normal se inicia cuando se
rompe la barrera de células endoteliales normales que recubre todos los vasos
sanguíneos. Cuando las células endoteliales se desprenden después de una lesión
vascular, la sangre que fluye queda expuesta a proteínas subendoteliales
vasculares (principalmente colágeno). Las plaquetas circulantes se adhieren
rápidamente al colágeno expuesto; se activan y experimentan un cambio dramático
en su forma y secretan su contenido de gránulos; y recluta plaquetas
adicionales al sitio de la lesión, formando un agregado de plaquetas o un tapón
hemostático que detiene temporalmente el flujo de sangre fuera del vaso dañado.
Este proceso a menudo se denomina hemostasia primaria y se inicia a los pocos
segundos de la lesión.
Al mismo tiempo, se activa el sistema de coagulación,
lo que lleva a la formación de una red de fibrina que envuelve y estabiliza el
tapón de plaquetas. La coagulación de la sangre se inicia por la interacción
del flujo sanguíneo con el factor tisular e implica una serie de reacciones
proteolíticas vinculadas. El evento de coagulación final es la generación de
suficiente trombina para convertir el fibrinógeno plasmático en fibrina. Este
proceso se ha denominado hemostasia secundaria y se completa varios minutos
después de la lesión. Horas o días más tarde, el tapón definitivo de fibrina/plaquetas
se disuelve lentamente por la vía fibrinolítica para que se pueda restablecer
el flujo sanguíneo en el vaso recién endotelizado.
La trombosis es el equivalente patológico de la
hemostasia normal y se la denomina hemostasia en el momento o lugar
equivocados. Así como es imposible desarrollar fármacos inmunosupresores que no
perturben el proceso inmunitario e inflamatorio normal, los fármacos diseñados
para prevenir o limitar la formación de trombos aumentan inevitablemente el
riesgo de hemorragia. En la trombosis arterial, el evento desencadenante, más
que la lesión vascular, es la patología dentro del endotelio o subendotelio
vascular. La ruptura de una placa aterosclerótica es la patología arterial más
frecuente. En la trombosis venosa, puede haber una combinación de generación
excesiva de trombina y lesión endotelial más sutil.
Las plaquetas son de importancia crítica para la
hemostasia en la microvasculatura y en la piel y las membranas mucosas. Por lo
tanto, los trastornos plaquetarios tienden a causar hemorragia principalmente
en estas áreas. Por el contrario, la vía de la coagulación es necesaria para la
hemostasia óptima en los vasos más grandes y en las articulaciones y los
músculos, por lo que las deficiencias conducen a la hemorragia tardía profunda
característica y hemartrosis.
La coagulación se dividió inicialmente en extremidades
intrínseca o dependiente del contacto y extrínseca o dependiente del factor
tisular. Ahora está claro que esta separación es artificial y no refleja cómo
proceden las reacciones in vivo. En la actualidad, existe consenso en que la
coagulación basal está impulsada por la formación de un complejo factor
tisular-VIIa, que activa los factores IX y X con igual eficacia. Además, las
trazas de trombina generadas por estas reacciones pueden retroalimentarse y
activar el factor XI. El papel de la activación por contacto a través del
factor XII (factor de Hageman) en la coagulación normal es ahora menos claro.
Evaluación Clínica y Pruebas de Coagulación
Los puntos clave a cubrir en la historia de cualquier
paciente con sospecha de trastorno hemostático se pueden resumir en estas siete
preguntas:
1. ¿Ha sangrado
el paciente en múltiples ocasiones y por múltiples sitios?
2. ¿Alguno de
los sangrados ha sido lo suficientemente grave como para requerir transfusión
de sangre?
3. ¿Qué tipos
de cirugía o trauma precipitaron el sangrado?
4. ¿Cuánto
tiempo después de la lesión ocurrió el sangrado?
5. ¿Existe un
patrón o ubicación específica del sangrado?
6. ¿Hay
antecedentes familiares de sangrado anormal y cuál es el patrón de la herencia?
7. ¿Qué
medicación, si alguna, toma el paciente?
Los elementos clave del examen físico incluyen:
1. Cualquier
evidencia de sangrado de piel o membranas mucosas (petequias, equimosis)
2. Evidencia de
hinchazón, acumulación de líquido, rango limitado de movimiento, o
engrosamiento sinovial de una articulación: caderas, rodillas, tobillos,
hombros, codos son los más afectados en hemofilia A y B (deficiencia de
factores VIII y IX)
3. Hematomas en
tejido subcutáneo profundo o músculo así como sangrado en la cabeza, las vías
respiratorias o el retroperitoneo que está fuera de proporción con el trauma
conocido o patología
4. Lesiones
vasculares como los hemangiomas nasales y labiales visto en la enfermedad de
Osler-Weber-Rendu o la piel anormal, laxitud e hiperextensibilidad articular
observadas en síndrome de Ehlers-Danlos.
Pruebas de detección de hemostasia
Las pruebas de detección básicas deben incluir lo
siguiente:
1. Conteo
sanguíneo completo (CBC)
2. Tiempo de
protrombina (PT), tiempo de tromboplastina parcial (PTT)
3. Estudios de
mezcla para descartar un inhibidor y/o identificar anomalías del factor si se
prolonga el PT o el PTT
4.
Nivel/actividad del factor von Willebrand (vWF) en pacientes con sospecha de
defecto hemostático primario por antecedentes
5. Agregación
plaquetaria en pacientes con sospecha de herencia, defecto hemostático primario
adquirido o inducido por fármacos
Trastornos Específicos
Enfermedad de von Willebrand
La enfermedad de von Willebrand (vWD) (von Willebrand
disease), es una de los más comunes trastornos hereditarios, que afectan a una
estimación de 1 de cada 100 personas. Muchos pacientes se ven mínimamente
afectados y pueden vivir toda su vida sin hemorragias adversas. vWD es un
trastorno autosómico dominante que afecta a hombres y mujeres con igual
frecuencia, presentándose como sangrado posquirúrgico o procedimientos
dentales, menorragia o moretones fáciles. El vWF (von Willebrand factor), es
una proteína plasmática heterogénea muy grande sintetizada en células
endoteliales y megacariocitos y secretada al plasma, así como al subendotelio
vascular. Se almacena en organelas endoteliales únicas llamadas cuerpos de
Weibel Palade o en los gránulos alfa de las plaquetas. vWF tiene dos funciones
principales: estabilizar la adhesión plaquetaria a la pared del vaso bajo
condiciones de alto flujo o “shear stress” uniéndose al colágeno y el complejo
GpIb plaquetario/IX/V, y servir como transportador intravascular de la proteína
antihemofílica factor VIII (figura 1).

Figura 1. Este esquema representa las relaciones entre
el factor de von Willebrand (vWF), con
las plaquetas y el endotelio.
La gran mayoría (85%) de los pacientes con vWD tienen la
enfermedad de tipo 1, causada por mutaciones sin sentido que perturban el
ensamblaje de multímeros (Tabla 1). Ellos, tienen una disminucion paralela en
el antígeno de vWF, la actividad de vWF medida como actividad del cofactor de
ristocetina y el factor VIII. Los niveles de vWF están influenciados por un
número de estados fisiológicos/patológicos o genes adicionales. Por ejemplo, la
inflamación aguda o crónica puede elevar el nivel de vWF, mientras que el
hipotiroidismo reduce el nivel de vWF. El entorno hormonal único presente
durante el embarazo puede normalizar completamente el nivel de vWF, lo que
permite un fácil trabajo de parto. La proteína vWF contiene moléculas del grupo
sanguíneo ABO que influencian la tasa de clearence del vWF del plasma. El vWF
tipo O se elimina más rápidamente, los tipos A y B menos y el grupo AB el más
lento. Por lo tanto, los pacientes de tipo O tienen los niveles plasmáticos más
bajos de vWF y es más probable que tengan sangrado cuando han heredado un alelo
vWD mutante.

Tabla 1. Caracterización en laboratorio de los tipos
de enfermedad de von Willebrand.
La mayoría de los pacientes restantes tienen vWD tipo
2 caracterizada por mutaciones específicas en el dominio vWF A1 que hacen que
la molécula sea anormalmente sensible a la degradación proteolítica (enfermedad
tipo 2a) o parcialmente activada y uniéndose continuamente a las plaquetas
circulantes (tipo 2b). Algunos pacientes raros con mutaciones que inactivan el
sitio en el dominio A1 que se une a GpIb (enfermedad tipo 2M).
Algunos pacientes tienen un trastorno que se ha
denominado hemofilia autosomal y tienen una mutación en la región del vWF que
se une y estabiliza el factor VIII (enfermedad tipo 2N). Cuando un alelo tipo
2N se combina con un alelo mutante tipo 1, el paciente heterocigoto doble
resultante puede tener un muy bajo nivel de factor VIII y se presentan con
hemartrosis que imitan la hemofilia clásica. Debido a que la función adhesiva
plaquetaria del vWF está conservado, no hay sangrado de las mucosas. Un patrón
de herencia autosómico puede proporcionar la clave para el diagnóstico y
distinguir esta condición de la hemofilia A clásica.
Hay un pequeño número de pacientes con enfermedad tipo
3, que es causado por grandes deleciones en el gen vWF. Estos pacientes han
heredado dos alelos anormales y tienen graves sangradoa de por vida sin vWF
detectable en su plasma.
Hemofilia adquirida y enfermedad de von Willebrand
En raras ocasiones, los pacientes con hemostasia
perfectamente normal durante toda su vida pueden desarrollar un defecto
hemostático grave causado por la adquisición de un inhibidor de anticuerpos
contra un factor de coagulación en particular, así como por la adsorción de un
factor de coagulación en la superficie de un tumor o una proteína anormal. Estos
trastornos presentan desafíos particulares y, en ocasiones, pueden causar
hemorragias muy graves, a veces letales.
La hemofilia adquirida generalmente es causada por un
anticuerpo contra el factor VIII. Se observa en pacientes con un trastorno
autoinmune como el lupus sistémico, en mujeres embarazadas y en personas de
edad avanzada sanas. La presentación en pacientes mayores sanos es el evento
más común. Los pacientes requieren apoyo intensivo con concentrados de factor
VIII y, más recientemente, factor VIIa recombinante. Con la terapia
inmunosupresora usando agentes como rituximab (Rituxan), junto con el paso del
tiempo, la mayoría de estos inhibidores desaparecerán y los pacientes se
recuperarán por completo.
El primer ejemplo de adsorción de un factor de
coagulación que causa una deficiencia adquirida, es la interacción del factor X
con la proteína amiloide en pacientes con amiloidosis primaria de cadena
ligera. Posteriormente, varios grupos han observado vWD adquirida debido a la
adsorción de vWF en las superficies tumorales. Esto es particularmente común en
pacientes con trastornos linfoproliferativos. La terapia eficaz requiere la
reducción de la masa tumoral.
Los pacientes con gammapatía monoclonal de significado
incierto pueden tener anticuerpos contra la proteína vWF y sangrado
significativo. Un número considerable de pacientes con macroglobulinemia de
Waldenstrom, mieloma y otros trastornos linfoproliferativos desarrollarán
anticuerpos anti-vWF y adquirirán vWD.
Finalmente, los pacientes con estenosis aórtica,
pacientes con dispositivos de asistencia ventricular y pacientes con trastornos
mieloproliferativos pueden desarrollar y luego proteolizar el vWF y desarrollar
vWD de leve a moderado.
Trastornos plaquetarios cualitativos
Los trastornos plaquetarios cualitativos son un grupo
heterogéneo de anormalidades que afectan muchos pasos diferentes en la adhesión
plaquetaria, señalización, empaquetamiento de gránulos y secreción y
agregación. Algunos trastornos son bastante comunes, mientras que otros son
extremadamente raros, y uno puede pasar toda una carrera en una práctica de
atención primaria o subespecialidad sin ver a un paciente con uno de estos
trastornos. Algunas anormalidades ocurren de forma aislada, mientras que otras
son una manifestación de un trastorno sistémico multiorgánico. Es conveniente
vincular los trastornos a pasos específicos en la función plaquetaria como se
muestra en la Fig.2.

Figura 2. Función plaquetaria.
Los trastornos de la membrana plaquetaria que afectan
a la adhesión o agregación, dos pasos críticos en la función plaquetaria, son
el resultado de la actividad cooperativa entre una glicoproteína de membrana y
una glicoproteína plasmática. La interacción del vWF con el complejo GpIb/IX/V
facilita la adhesión plaquetaria, mientras que la unión del fibrinógeno a la
GpIIb/IIIa regula la agregación plaquetaria. Los pacientes raros con mutaciones
en los polipéptidos GpIb aob o GpIX no pueden sintetizar el complejo GpIb/IX/V,
una condición llamada síndrome de Bernard-Soulier. Se caracteriza por plaquetas
anormalmente grandes, trombocitopenia de leve a moderada e incapacidad para
soportar la adhesión dependiente de vWF. Es un rasgo autosómico recesivo y
causa sangrado de por vida. De manera similar, los pacientes con mutaciones en
los polipéptidos GpIIb o GpIIIa no logran sintetizar el complejo plaquetario
GpIIb/IIIa y tienen plaquetas que no pueden unirse al fibrinógeno ni agregarse.
Este trastorno, llamado trombastenia de Glanzmann, también es un rasgo
autosómico recesivo. Se diferencia del síndrome de Bernard-Soulier en que los
pacientes tienen un recuento de plaquetas normal y plaquetas de tamaño normal.
Al igual que los pacientes de Bernard Soulier, también tienen hemorragia grave
y recurrente de por vida. En ambos casos, las transfusiones de plaquetas
repetidas pueden conducir a la aloinmunización debido a los anticuerpos
dirigidos contra las proteínas que faltan, lo que puede limitar la eficacia de
las transfusiones de plaquetas en el futuro.
Se han identificado pacientes con defectos selectivos
en el transporte y envasado de materiales en gránulos de plaquetas. Los
pacientes con enfermedad de cuerpo denso o acumulación delta tienen niveles
bajos de trifosfato de adenosina, difosfato de adenosina (ADP) gránulos, calcio
y serotonina y agregación plaquetaria secundaria defectuosa. Por el contrario,
los pacientes con enfermedad de los gránulos alfa o del pool de almacenamiento
alfa tienen una agregación normal o casi normal. Los pacientes con enfermedad
combinada alfa/delta tienen plaquetas que tienen la apariencia de queso suizo
con múltiples orificios que representan la membrana limitante de los gránulos
vacíos. Tienen un defecto hemostático y también pueden desarrollar ibrosis de
la médula cuando proteínas como el factor de crecimiento derivado de las
plaquetas se filtran de los megacariocitos y estimulan el crecimiento de los
fibroblastos de la médula.
Los pacientes con albinismo oculocutáneo y los
pacientes con el síndrome de Chédiak-Higashi, que también pueden ser albinos
parciales, tienen un defecto de empaquetamiento de gránulos generalizado que se
extiende a las plaquetas y se presenta como una enfermedad delta pool de
almacenamiento. Los pacientes con síndrome de Hermansky-Pudlak tienen
enfermedad delta pool de almacenamiento y, a menudo, desarrollan fibrosis
pulmonar grave. Muchos de estos pacientes terminan requiriendo oxigenoterapia
continua y eventuales trasplantes de pulmón.
Se han identificado pacientes con mutaciones en el
receptor P2Y12ADP y en algunas de las moléculas importantes de señalización
intraplaquetaria. Una mutación en una isoforma de miosina, MyH9, causa la
anomalía de May-Hegglin, que se caracteriza por plaquetas muy grandes,
trombocitopenia moderada y cuerpos de Dohle en sus leucocitos, pero sin defecto
hemostático.
En la práctica clínica, las anomalías plaquetarias más
comunes son las causadas por la administración de medicamentos antitrombóticos.
La aspirina es el fármaco administrado con mayor frecuencia e induce un defecto
hemostático leve. Debido a que inactiva irreversiblemente la ciclooxigenasa
plaquetaria, una sola dosis puede perturbar la hemostasia durante 5 a 7 días.
Otros fármacos antiinflamatorios no esteroideos (AINE), como el naproxeno o el
ibuprofeno, son inhibidores reversibles transitorios de la ciclooxigenasa y
rara vez provocan hemorragia clínica. De mucha más importancia es su
competencia con la aspirina por la unión de la ciclooxigenasa. La ingestión
simultánea de naproxeno sódico o ibuprofeno y aspirina bloqueará el efecto
cardiovascular deseado de la aspirina y es una de las principales causas de la
resistencia a la aspirina. Se debe indicar a los pacientes que primero tomen
aspirina y que esperen al menos 30 minutos antes de tomar un AINE.
El clopidogrel y el prasugrel son inhibidores de P2Y12
que bloquean la agregación inducida por ADP. Son profármacos cuyos metabolitos
activos son inhibidores irreversibles, por lo que su efecto también es
prolongado. Otros dos medicamentos populares, la integrelina y el abciximab
(Rheo Pro), se unen al complejo GpII/IIIa y bloquean la unión del ibrinógeno
plaquetario y la agregación plaquetaria. Integrelin tiene una vida media
biológica corta y puede revertirse rápidamente al detener su infusión. El
efecto de abciximab puede persistir durante varios días.
Hemofilia A
Aunque se han descrito pacientes con deficiencias en
cada una de las proteínas de la coagulación conocidas, predominan tres
enfermedades y representan más del 90% de los pacientes con trastornos
hereditarios de la coagulación: deficiencias en los factores VIII, IX y XI.
También se conocen como hemofilias A, B y C. La deficiencia de los factores
VIII y IX son trastornos ligados al cromosoma X que afectan principalmente a
los hombres, mientras que la deficiencia del factor XI es un trastorno
autosómico recesivo que puede afectar tanto a hombres como a mujeres.
La deficiencia del factor VIII se produce en 1 de cada
10.000 nacimientos de varones y provoca hemorragias recurrentes de por vida en
los tejidos blandos, los músculos y, lo que es más importante, hemorragias
articulares o hemartrosis. Existe una estrecha relación entre el nivel de
factor VIII y la gravedad del sangrado. Los pacientes con <1% de actividad
tienen una enfermedad grave con hemorragia frecuente que pone en peligro la vida.
Los pacientes con 1% a 5% de actividad tienen enfermedad moderada con sangrado
semanal o incluso mensual. Los pacientes con niveles superiores al 5% tienen
una enfermedad más leve con sangrado poco frecuente.
El tratamiento de los hemofílicos ha mejorado constantemente.
En la actualidad, muchos niños y adolescentes reciben terapia profiláctica
varias veces a la semana y tienen pocas hemorragias graves; casi todos los
niños y adultos se autoadministran concentrados de factor de coagulación en el
hogar a demanda con una supervisión médica mínima; y la mayoría de los
pacientes usan concentrados de factor recombinante altamente purificados que
están libres de todos los virus conocidos. Los avances recientes en las
opciones de tratamiento para la hemofilia A pueden revolucionar los enfoques
actuales a través del desarrollo de un mimético del factor VIII (emicizumab).
Shima y colegas (2016) demostraron recientemente que el emicizumab, un fármaco
que funciona como una réplica conformacional del factor VIII al unirse a los
factores IX y X en un complejo generador de trombina, tiene eficacia en el
tratamiento de la hemofilia grave. Ciertamente, esto haría avanzar el campo no
solo al proporcionar un enfoque diferente a la terapia, sino también al ofrecer
un tratamiento que se asocia con menos anticuerpos neutralizantes del factor
VIII, como se analiza más adelante.
Aunque la esperanza de vida de un paciente con
hemofilia es casi normal y muchos pacientes tienen pocas articulaciones
dañadas, existen problemas de salud sin resolver, como el aumento de la
incidencia de hipertensión y el enorme gasto de una terapia óptima. Quizás la
complicación más temida de la hemofilia en la actualidad es el desarrollo de un
inhibidor del factor VIII. Esto ocurre en 15% a 20% de los pacientes y complica
la terapia y reduce la calidad de vida del paciente. Recientemente, Peyvandi y
colaboradores (2016) demostraron que la incidencia acumulada de desarrollo de
inhibidores en niños con hemofilia A grave fue mayor en los que recibieron
productos de factor VIII recombinante en comparación con los que recibieron
factor VIII derivado de plasma que contenía vWF. Este estudio tiene
implicaciones para la toma de decisiones clínicas con respecto a la terapia de
reemplazo en hemofílicos con enfermedad grave, aunque la tasa de desarrollo de
inhibidores aún es baja.
Hemofilia B y C
Casi todo lo escrito anteriormente sobre la
deficiencia de factor VIII es válido para la deficiencia de factor IX. Es menos
común, aparece en 1 de cada 50 000 nacimientos, y la proteína tiene una vida
media plasmática más prolongada, por lo que las infusiones son menos
frecuentes. De lo contrario, las enfermedades son casi idénticas.
Sin embargo, la deficiencia del factor XI es bastante
distinta. Primero, es autosómico recesivo y generalmente se presenta como
sangrado postoperatorio. Es más común en las poblaciones judías Ashkenazi.
Además, la correlación entre el nivel de factor y el sangrado no es muy fuerte
por razones desconocidas.
Finalmente, en los Estados Unidos, el único tratamiento
disponible es la infusión de plasma fresco congelado porque el concentrado de
factor XI no ha sido aprobado debido a la preocupación por un mayor riesgo
trombótico.
Hemofilia adquirida y enfermedad de von Willebrand
En raras ocasiones, los pacientes con hemostasia
perfectamente normal durante toda su vida pueden desarrollar un defecto
hemostático grave causado por la adquisición de un inhibidor de anticuerpos
contra un factor de coagulación en particular, la adsorción de un factor de
coagulación en la superficie de un tumor o una proteína anormal. Estos
trastornos presentan desafíos particulares y, en ocasiones, pueden causar
hemorragias muy graves, a veces letales.
La hemofilia adquirida generalmente es causada por un
anticuerpo contra el factor VIII. Se observa en pacientes con un trastorno
autoinmune como el lupus sistémico, en mujeres embarazadas y en personas de
edad avanzada sanas. La presentación en pacientes mayores sanos es el evento
más común. Los pacientes requieren apoyo intensivo con concentrados de factor
VIII y, más recientemente, factor VIIa recombinante. Con la terapia
inmunosupresora usando agentes como rituximab (Rituxan), junto con el paso del
tiempo, la mayoría de estos inhibidores desaparecerán y los pacientes se
recuperarán por completo.
El primer ejemplo de adsorción del factor de
coagulación que causa una deficiencia adquirida es la interacción del factor X
con la proteína amiloide en pacientes con amiloidosis primaria de cadena
ligera. Posteriormente, varios grupos han observado vWD adquirida debido a la
adsorción de vWF en las superficies tumorales. Esto es particularmente común en
pacientes con trastornos linfoproliferativos. La terapia eficaz requiere la
reducción de la masa tumoral.
Los pacientes con gammapatía monoclonal de significado
incierto pueden tener anticuerpos contra la proteína vWF y sangrado
significativo. Un número considerable de pacientes con macroglobulinemia de
Waldenstrom, mieloma y otros trastornos linfoproliferativos desarrollarán
anticuerpos anti-vWF y adquirirán vWD.
Finalmente, los pacientes con estenosis aórtica,
pacientes con dispositivos de asistencia ventricular y pacientes con trastornos
mieloproliferativos pueden desarrollar y luego proteolizar el vWF y desarrollar
vWD de leve a moderado.
Trombocitopenia inmune
La trombocitopenia inmune, anteriormente llamada
púrpura trombocitopénica idiopática (PTI), es el trastorno autoinmune más
común. En los niños pequeños, es un trastorno transitorio que sigue a una
infección viral. En los adultos, la PTI suele ser un problema crónico que
afecta tres veces más a las mujeres sanas que a los hombres. Los pacientes rara
vez pueden tener otros fenómenos autoinmunes. Por ejemplo, la aparición
simultánea o secuencial de anemia hemolítica autoinmune y trombocitopenia se
conoce como síndrome de Evans. Aunque la PTI rara vez es fatal, puede causar
hemorragia mucocutánea recurrente y, en ocasiones, grave e intracerebral
ocasional. El antígeno diana más frecuente es el complejo plaquetario
GpIIb/IIIa. Un pequeño número de pacientes tiene anticuerpos contra el complejo
GpIb/IX/V u otras proteínas de la superficie celular de las plaquetas. En la
mayoría de los casos, los anticuerpos actúan como opsoninas y aumentan la
eliminación de plaquetas de la circulación sin alterar la función plaquetaria.
Ocasionalmente, el anticuerpo puede alterar la unión del fibrinógeno y los
pacientes tendrán trombocitopenia y disfunción plaquetaria que simula la
enfermedad de Glanzmann. Ha habido múltiples intentos de desarrollar pruebas de
laboratorio para autoanticuerpos plaquetarios en pacientes con PTI. Ninguna de
las pruebas ha tenido éxito por innumerables razones, incluido un alto nivel de
IgG de fondo en la superficie de las plaquetas y la presencia de receptores Fc,
que pueden unirse a inmunoglobulinas o complejos inmunitarios de manera no
específica. El paciente típico con PTI presenta antecedentes de hematomas
fáciles, sangrado mucocutáneo y, si el recuento de plaquetas es lo
suficientemente bajo, petequias, que surgen del movimiento de glóbulos rojos a
través de capilares con fugas hacia la piel. La mayoría de los pacientes no
tienen hallazgos físicos patognomónicos ni pruebas de laboratorio, y la PTI
sigue siendo un diagnóstico de exclusión. En contraste con los pacientes que
tienen anemia hemolítica autoinmune, los pacientes con PTI tienen un bazo de
tamaño normal. Por lo general, aparte de la trombocitopenia, el hemograma es
normal, aunque algunos pacientes pueden tener linfocitos atípicos, lo que
sugiere una infección viral reciente. Aquí hay un debate sobre lo que
constituye un estudio adecuado para la PTI. La mayoría de los hematólogos han
dejado de realizar exámenes de médula ósea en pacientes con PTI a menos que se
sospeche una anomalía hematológica más global. Debido a la asociación con la
enfermedad autoinmune, el estudio suele incluir una prueba de anticuerpos
antinucleares, que suele ser normal. Muchos médicos ordenan rutinariamente la
prueba del VIH en todos los pacientes que son sexualmente activos, mientras que
otros lo ordenan solo si el paciente ha tenido un comportamiento de alto
riesgo. Los paneles serológicos para toxoplasmosis, citomegalovirus y otros
trastornos virales rara vez son positivos y no se recomiendan a menos que estén
clínicamente indicados en el momento de la presentación. La PTI crónica se
define como la trombocitopenia que ha estado presente durante al menos 3 meses.
La probabilidad de una etiología viral o una remisión espontánea es
extremadamente baja después de 3 meses. Durante muchos años, la terapia inicial
estándar ha sido la administración de grandes dosis de glucocorticoides, por lo
general 50 mg de prednisona o equivalente al día. En la mayoría de los
pacientes, el recuento de plaquetas vuelve a la normalidad después de varias
dosis de prednisona, pero cae a los valores previos al tratamiento a medida que
se reduce la dosis de esteroides. Si el recuento permanece bajo después de
varios meses de tratamiento con prednisona, el tratamiento de segunda línea
bien establecido es la esplenectomía. En la mayoría de los centros grandes,
este es un procedimiento laparoscópico con una morbilidad y mortalidad mínimas.
Los pacientes son inmunizados contra organismos encapsulados como neumococo,
meningococo y Haemophilus influenzae que se eliminan principalmente en el bazo.
La única infección restante que empeora con la esplenectomía es la babesiosis.
En adultos, el bazo parece ser prescindible y la función inmunológica se
conserva en gran medida. La esplenectomía eleva el recuento de plaquetas a la
normalidad en aproximadamente el 70% de los pacientes con PTI. A los pacientes
que fallan en la esplenectomía y tienen recuentos de plaquetas peligrosamente
bajos (<50 000/μL) generalmente se les administran medicamentos
inmunosupresores azatioprina (Imuran) o ciclofosfamida oral. Recientemente, el
fármaco preferido es el anticuerpo monoclonal antiCD20 rituximab. Inducirá una
remisión en el 70% de los pacientes en los que han fracasado los
corticosteroides y la esplenectomía, pero puede requerir un segundo curso de
tratamiento dentro de un año en el 25% de los respondedores iniciales. Aunque
la tasa de complicaciones es baja, las infecciones oportunistas son un problema
potencial; varios pacientes han desarrollado leucoencefalopatía multifocal
progresiva después del tratamiento con rituximab, por lo que se recomienda
precaución. Existe un gran deseo entre los pacientes y los médicos tratantes de
evitar la esplenectomía. Un nuevo enfoque es la administración de pulsos de
dexametasona en dosis muy altas durante 4 días al mes. Después de varios meses
de terapia, un pequeño porcentaje de pacientes entra en remisión. La tasa de
remisión puede aumentar cuando los pacientes reciben pulsos de dexametasona y
cuatro dosis de rituximab como terapia inicial. Aunque este régimen puede inducir
la remisión
Trombocitopenia inducida por heparina
La heparina es la causa más común de trombocitopenia
en pacientes hospitalizados y afecta del 15% al 20% de los pacientes que
reciben heparina no fraccionada. La trombocitopenia inducida por heparina (HIT)
es causada por un anticuerpo dirigido contra un complejo de heparina y la
proteína neutralizante de heparina, el factor plaquetario 4 (PF4). El complejo
de anticuerpos heparina PF4 se une al receptor Fc plaquetario, lo que induce
tanto la activación y secreción plaquetaria como la trombocitopenia. El
espectro de HIT varía desde pacientes con trombocitopenia leve no progresiva
hasta pacientes que desarrollan trombocitopenia profunda y un paciente
ocasional que desarrolla trombosis potencialmente mortal a pesar de estar
totalmente anticoagulado. Existe un mayor riesgo de formación de trombos en
todos los pacientes con HIT, que persiste durante varios meses después de
suspender la heparina.

Figura 3: Mecanismo de la trombocitopenia inducida por
heparina
Algunas personas producen anticuerpos IgG dirigidos
contra el complejo heparina PF4. Los receptores plaquetarios Fc se unen al
complejo inmunitario anticuerpo-heparina-PF4. Esto conduce a la activación
plaquetaria y la liberación de micropartículas, que contribuyen a la trombosis.
La trombocitopenia ocurre por dos mecanismos: eliminación de plaquetas con IgG
unida por macrófagos esplénicos y consumo de plaquetas causado por la formación
de trombos. PF4 también puede unirse al sulfato de heparán en las células
endoteliales vasculares; la unión posterior del anticuerpo patológico a este
complejo de PF4-heparán sulfato puede lesionar el endotelio, lo que promueve
aún más la trombosis.
La HIT se diagnostica mediante una combinación de
observación clínica y pruebas de laboratorio juiciosas. Las cuatro
características clave son el grado de trombocitopenia, el momento de la
trombocitopenia, la presencia de trombosis concomitante y la ausencia de otras
causas obvias de trombocitopenia. Una caída de más del 50 % del recuento de
plaquetas desde que se inició la heparina con un nadir >20 000/μL; aparición
de trombocitopenia de 5 a 14 días después de comenzar con la heparina, 48 horas
si se expuso previamente a la heparina dentro de los 30 días; y la trombosis
nueva, la necrosis cutánea o la reacción anafiláctica a la infusión de heparina
se consideran fuertes predictores de TIH. Si se sospecha HIT, se debe solicitar
una prueba de inmunoabsorción ligada a enzimas (ELISA) de heparina PF4. La
prueba tiene una sensibilidad reportada del 95% y por lo tanto un alto valor
predictivo negativo. La limitación de la prueba es que no distingue entre los
anticuerpos IgM e IgA o los anticuerpos IgG que causan la activación
plaquetaria. La especificidad informada del 50 % se puede mejorar observando la
densidad óptica (DO) de la prueba ELISA. Es más probable que una OD >1.00
sea causada por un anticuerpo IgG patológico. Se están introduciendo pruebas
más nuevas que utilizan antisueros específicos de IgG que deberían aumentar la
especificidad de la prueba. Un segundo conjunto de pruebas que miden la
activación plaquetaria, como el ensayo de liberación de serotonina, puede
identificar los anticuerpos que tienen más probabilidades de causar HIT y se
dice que tienen una sensibilidad y especificidad >90 %. La prueba es
bastante especializada, no está ampliamente disponible y solo se puede realizar
una o dos veces por semana, incluso en grandes laboratorios de referencia.
Una vez que se identifica HIT, la infusión de heparina
debe suspenderse inmediatamente y los pacientes deben cambiar a un inhibidor
directo de la trombina. Los dos fármacos más utilizados para tratar la TIH son
el argatrobán, un derivado de molécula pequeña de la l-arginina con una
semivida plasmática de 45 minutos y la lepirudina (hirudina recombinante), que
tiene una semivida de 2 horas. Ambos medicamentos se administran por infusión
intravenosa y se controlan midiendo el PTT. Cuando el recuento de plaquetas
vuelve a >150 000/μl, los pacientes pasan a warfarina, que se continúa
durante 30 días en pacientes sin trombosis y de 3 a 6 meses en pacientes con
trombocitopenia inducida por heparina con trombosis. La incidencia de HIT
debería disminuir y eventualmente desaparecer a medida que se introducen nuevas
formas de heparina que son menos inmunogénicas. Por ejemplo, la incidencia de
HIT es <1% para las heparinas de bajo peso molecular como la enoxaparina
(Lovenox) o la dalteparina (Fragmin). Solo se han informado unos pocos casos de
TIH en pacientes que recibieron el pentasacárido sintético fondaparinux
(Arixtra). Dada la eficacia y seguridad de las nuevas heparinas, la heparina no
fraccionada probablemente debería reservarse para pacientes que requieren una
titulación minuto a minuto de la dosis de heparina y una pronta reversibilidad.
La heparina no fraccionada solo debe ser necesaria para cateterismo cardíaco,
derivación cardiopulmonar, en unidades de cuidados intensivos y, quizás, en
pacientes con insuficiencia renal.
Púrpura trombocitopénica trombótica
La púrpura trombocitopénica trombótica (PTT) es un
trastorno relativamente raro caracterizado por trombocitopenia, anemia
hemolítica microangiopática, diversos grados de insuficiencia renal y síntomas
neurológicos fluctuantes. La mayoría de los pacientes con PTT esporádica tienen
una deficiencia adquirida de ADAMTS13, una enzima metaloproteasa plasmática que
remodela el vWF secretado por las células endoteliales. En ausencia de esta
enzima, los multímeros supergrandes de vWF interactúan con las plaquetas
circulantes y forman los trombos hialinos característicos de la PTT. Aunque hay
pacientes raros que tienen una deficiencia congénita en ADAMTS13, la mayoría de
los pacientes con deficiencia adquirida tienen un inhibidor de autoanticuerpos.
Los pacientes que desarrollan TTP después de un trasplante de células madre o
de la ingestión de fármacos tienen niveles normales de ADAMTS13 y pueden tener
daño o disfunción endotelial que induce la liberación de grandes cantidades de
multímeros grandes (Fig. 4).
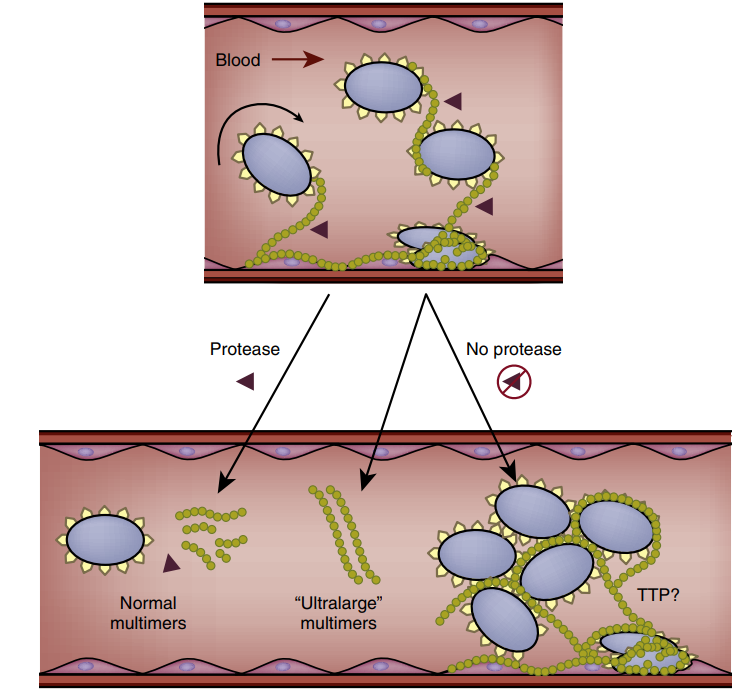
Figura 4. TTP. Rol esquemático del factor de von
Willebrand factor y la adhesión plaquetaria.
Los pacientes con trombocitopenia de inicio repentino,
anemia, niveles elevados de nitrógeno ureico y creatinina en sangre y anomalías
neurológicas (por lo general, niveles fluctuantes de conciencia o hallazgos
focales fluctuantes) son buenos candidatos para la TTP. El frotis de sangre
debe mostrar la presencia de esquistocitos, mientras que los parámetros de
coagulación, incluidos los niveles de PT, PTT, fibrinógeno y dímero D, son
normales. La lactato deshidrogenasa (LDH) elevada es una característica
cardinal. La sangre debe enviarse a un laboratorio de referencia para la
actividad de ADAMTS13 y los niveles de inhibidor, aunque es posible que los
resultados no estén disponibles durante varios días o una semana.
La mejor terapia para la PTT es la plasmaféresis
intensiva acompañada de infusión de plasma fresco congelado. Aunque la terapia
se derivó empíricamente, es racional. La plasmaféresis puede eliminar
anticuerpos o complejos anticuerpo-enzima, mientras que la infusión de plasma
reemplaza a ADAMTS13. Una vez iniciada, la plasmaféresis diaria debe
continuarse hasta que los síntomas neurológicos hayan disminuido y la
creatinina vuelva a la normalidad junto con el recuento de plaquetas y la LDH.
Aproximadamente el 20% de los pacientes pueden sufrir una recaída
inmediatamente después de detener la plasmaféresis y pueden requerir un nuevo
tratamiento. Dentro de un año del tratamiento inicial, el 20% de los pacientes
pueden recaer y requerir plasmaféresis adicional. Antes del advenimiento de la
plasmaféresis y el reemplazo de plasma, la mortalidad de la PTT era cercana al
100%. Ahora se ha reducido a 10% a 15%.
Hay una larga lista de terapias que no han sido
efectivas en la PTT. Incluyen medicamentos antiplaquetarios, esplenectomía y
algunos de los medicamentos inmunosupresores más antiguos, como la prednisona y
la azatioprina. Recientemente ha habido evidencia de que el anticuerpo
monoclonal anti-CD20 rituximab puede ser beneficioso en la TTP y ciertamente
debe probarse en pacientes con formas recurrentes del trastorno. Además, se ha
desarrollado una inmunoglobulina humanizada de dominio único variable anti-vWF
llamada caplacizumab y ha generado resultados inicialmente prometedores.
Peyvandi y colaboradores (2016) publicaron recientemente los resultados de un
estudio controlado de fase 2 en pacientes con TTP adquirida que recibieron
caplacizumab y demostraron una resolución más rápida de los episodios agudos de
TTP en comparación con el grupo de placebo.
Los fármacos más comunes que causan PTT son las
tienopirimidinas ticlopidina y su derivado cercano clopidogrel, que se unen al
receptor plaquetario P2Y12 (ADP). Aunque son agentes antitrombóticos muy
eficaces, la incidencia de PTT tras la administración de ticlopidina fue tan
alta que el fármaco se retiró del mercado. Se ha descrito TTP tras la
administración de clopidogrel, pero teniendo en cuenta el uso generalizado de
este fármaco la incidencia es bastante baja. La mortalidad en pacientes con PTT
inducida por fármacos es mayor que en los casos esporádicos y se acerca al 50%.
Aunque se prescribe plasmaféresis, es menos claro que sea eficaz en este
subconjunto de TTP.
Coagulación intravascular diseminada
La coagulación intravascular diseminada (CID) es
causada por la activación no regulada de la vía de la coagulación. Se observa
con mayor frecuencia durante el trabajo de parto y el parto y en pacientes con
sepsis o malignidad. El desencadenante de la DIC puede ser la endotoxina de las
bacterias, un factor tisular u otros activadores de la coagulación y el
contacto de la sangre con superficies o membranas incompatibles. La coagulación
no regulada conduce a una generación excesiva de trombina, lo que provoca la
conversión rápida del fibrinógeno plasmático en fibrina. Las plaquetas quedan
atrapadas en los trombos de fibrina y los glóbulos rojos se ensartan en las
hebras de fibrina. La respuesta fibrinolítica al depósito masivo de fibrina en
la microcirculación conduce a anomalías adicionales de la coagulación.
Los hallazgos de laboratorio clásicos incluyen
trombocitopenia, anemia con esquistocitos en el frotis de sangre, PT y PTT
prolongados y fibrinógeno bajo. Los pacientes también tienen productos de
degradación de fibrinógeno/fibrina elevados. El ensayo más común en uso hoy en
día es el ensayo de dímero D, que utiliza un anticuerpo monoclonal específico
de fibrina para detectar productos de degradación de fibrina reticulada.
La DIC se diferencia fácilmente de la TTP, pero puede
ser más difícil distinguir la DIC de un estado fibrinolítico primario. La
fibrinólisis primaria es un evento raro que se observa con algunas neoplasias
malignas que tienen altas concentraciones de activadores fibrinolíticos, como
el carcinoma de próstata, o en pacientes con cirrosis avanzada que no eliminan
los activadores fibrinolíticos de la sangre. Aunque en teoría, los pacientes
con fibrinólisis deberían tener recuentos de plaquetas normales, ausencia de
esquistocitos y análisis de dímero D normales, en situaciones clínicas estas
distinciones pueden volverse borrosas, quizás debido a una combinación de CID y
fibrinólisis primaria, limitaciones en el análisis de dímero D , o los efectos
de la plasmina en las plaquetas.
Los pacientes con DIC pueden presentarse con trombosis
de vasos pequeños, a menudo en dedos, extremidades, piel o genitales, con
hemorragia fulminante en múltiples sitios o con alguna combinación de
hemorragia y trombosis. Las pacientes con DIC secundaria a sepsis parecen tener
una trombosis más prominente, mientras que las pacientes con DIC obstétrica
tienden a tener sangrado masivo incontrolable. Muchos pacientes con cáncer
tienen DIC crónica de bajo grado, pero pueden desarrollar una enfermedad más
activa si se someten a una resección del tumor u otra cirugía. Los pacientes
con tumores epiteliales que han hecho metástasis a los vasos sanguíneos pueden
desarrollar DIC fulminante e intratable.
El tratamiento de la CID varía según la manifestación
clínica. Los pacientes que tienen trombosis se tratan mejor con heparina. La
heparinización inmediata puede salvar la vida y prevenir la necrosis y
amputación subsiguiente del tejido. Los pacientes con sangrado generalmente se
tratan con plaquetas, glóbulos rojos y plasma fresco congelado para reemplazar
los factores de coagulación agotados. Después de esta reanimación inicial, el
siguiente paso más importante es intentar tratar la patología subyacente que
induce la CID. En mujeres embarazadas, las causas son placenta previa,
separación prematura de la placenta con coágulo retroplacentario, eclampsia
grave o retención de productos de la concepción. Con el parto del feto y la
placenta, la DIC puede desaparecer con bastante rapidez. El tratamiento de
gramnegativos u otras formas de sepsis puede ayudar a revertir la CID y detener
el sangrado. Aunque esto debería intentarse, no hay evidencia de que el
tratamiento de la DIC per se mejore el pronóstico en pacientes sépticos. Los
pacientes con tumor metastásico representan el mayor desafío porque es posible
que no exista una terapia eficaz para el tumor subyacente. Si el paciente
desarrolla DIC aguda en asociación con la cirugía, la terapia de reemplazo
puede ayudar a detener el sangrado aunque la DIC de bajo grado puede persistir.
La heparina se puede usar como complemento de la terapia de reemplazo si el
fibrinógeno y las plaquetas son persistentemente bajos a pesar del reemplazo
adecuado. Los pacientes con DIC deben ser seguidos de cerca con mediciones
seriadas del nivel de ibrinógeno, dímero D o cualquier otra medición de
fibrinógeno/productos de degradación de fibrina. El recuento de plaquetas puede
retrasarse con respecto a estos otros parámetros.
Estados hipercoagulables
Los pacientes con cáncer, insuficiencia cardíaca
congestiva, inmovilidad prolongada o pacientes sometidos a procedimientos
quirúrgicos tienen un mayor riesgo de trombosis. Los mecanismos no se conocen
bien y son multifactoriales. Cada vez se utiliza más la anticoagulación
profiláctica con heparina y/o warfarina en estos pacientes. Las más notables
son las marcadas reducciones en el tromboembolismo venoso posoperatorio en
pacientes ortopédicos con fractura de cadera o reemplazo de cadera o rodilla
después del uso universal de warfarina en el período perioperatorio. Se ha
identificado un grupo de rasgos genéticos que aumentan el riesgo de
tromboembolismo venoso y, en conjunto, pueden representar hasta el 70 % de los
pacientes con trombosis venosa profunda (TVP) recurrente o embolia pulmonar
(EP). Estas mutaciones también aumentan el riesgo de trombosis en pacientes que
están embarazadas, usan anticonceptivos orales, tienen cáncer o toman ciertos
medicamentos. Además, las mutaciones son lo suficientemente comunes como para
que los pacientes a menudo cohereden dos de los defectos, lo que aumenta el
riesgo de trombosis. Cada uno de estos trastornos, revisados más adelante,
tiene su propia historia natural, fisiopatología y respuesta al tratamiento
únicas (Tabla 2).

Tabla 2. Estados Hipercoagulables y Tipo de Trombosis.
1. Déficit de
antitrombina (AT): este fue el primero de este grupo de trastornos en ser
identificado. Es un rasgo autosómico dominante y ocurre en aproximadamente 1 de
cada 2000 individuos. Debido a que los pacientes solo tienen un alelo (gen)
afectado, solo tienen una deficiencia modesta en AT. No se han identificado
pacientes con dos alelos defectuosos; por lo tanto, creemos que la
homocigosidad es una condición embrionaria letal. La mayoría de los pacientes
con deficiencia de AT desarrollarán síntomas de TVP o EP antes de los 30 años.
Aunque hay concentrados de AT disponibles para la terapia de reemplazo, la
mayoría de los pacientes tienen solo una disminución modesta en el nivel de AT
y responden normalmente a la heparina. El raro paciente con una mutación sin
sentido que perturba la unión de heparina o la activación de AT inducida por
heparina o una mutación en el sitio activo de AT requerirá terapia de
reemplazo. Debido a que el riesgo de recurrencia es bastante alto, los
pacientes que tienen un evento trombótico inicial deben recibir anticoagulación
oral de por vida con warfarina o su equivalente. Los familiares de un paciente
con deifciencia conocida de AT deben hacerse la prueba y, si son portadores de
la mutación, deben evitar los anticonceptivos orales y recibir profilaxis con
cirugía electiva.
2. Deficiencia
de proteína S y C: estas dos proteínas, similares a los factores de coagulación
II, VII, IX y X, se sintetizan en el hígado y requieren una modificación
postraduccional (gamma carboxilación de ácidos glutámicos específicos) para la
actividad biológica. Cualquier cosa que perturbe la carboxilación gamma, como
la enfermedad hepática, la deficiencia de vitamina K o los anticoagulantes
orales de la clase de la warfarina, reducirá los niveles de las proteínas C y
S. La proteína C se une a la proteína trombomodulina de la superficie celular
endotelial, donde es activada por la trombina. La proteína C activada cataliza
la inactivación de los factores V y VIII, dos cofactores críticos en la vía de
la coagulación, junto con la proteína S y la trombomodulina de la proteína de
la superficie endotelial. Las deficiencias en las proteínas C y S son muy
comunes, con estimaciones para la proteína C tan comunes como 1 en 200
individuos. La mayoría de los individuos afectados son asintomáticos o
mínimamente afectados y es posible que nunca desarrollen trombosis venosa o
embolia. Sin embargo, los trastornos aumentan varias veces el riesgo de por
vida de DVT/PE. Los bebés con deficiencia homocigota de proteína C o S
desarrollan DIC fulminante justo después del nacimiento y requieren infusiones
de plasma de por vida para reemplazar la proteína C o S. Los padres de estos
niños gravemente afectados a menudo son completamente asintomáticos y solo
tienen una leve disminución de proteína C o S. Proteína C los niveles se
reducen en pacientes que toman warfarina. Debido a que la proteína C tiene una
vida media plasmática corta, durante el inicio de la terapia con warfarina su
nivel cae antes que los factores II, VII, IX o X, creando un estado protrombótico
transitorio. Es especialmente pronunciado en pacientes que comienzan con
warfarina y tienen deficiencia de proteína C. Se cree que este estado
protrombótico causa la rara complicación de necrosis cutánea inducida por
warfarina. Esta complicación grave es, afortunadamente, muy rara porque la
mayoría de los pacientes que comienzan con warfarina están tomando heparina y,
por lo tanto, están protegidos.
La proteína S actúa como cofactor de alto peso
molecular y forma un complejo con la proteína C y la trombomodulina para
facilitar la inactivación de los factores V y VIII. Existe en dos formas: una
fracción activa que está libre en el plasma y una fracción inactiva unida a una
globulina fijadora de esteroides. El embarazo y el uso de anticonceptivos
orales pueden aumentar el nivel de esta proteína y, por lo tanto, inducir o
exacerbar la deficiencia de proteína S. Esta reducción de la proteína S, cuando
se combina con otro defecto leve como el factor V Leiden o la mutación del gen
de la protrombina, puede explicar el aumento de la TVP/PE en el embarazo y en
las usuarias de anticonceptivos orales que antes eran asintomáticas.
3. Factor V
Leiden: su mutación R506Q está presente en el 5% de la población caucásica pero
es poco común en africanos, asiáticos y latinos. La mutación modifica uno de
los dos sitios sensibles a la proteasa en el factor V que son escindidos por la
proteína C activada y, por lo tanto, genera un exceso de trombina. A pesar de
mucha especulación, no sabemos cómo surgió y se propagó esta mutación
invariable. Portar la mutación aumenta el riesgo de por vida de TVP/EP
aproximadamente 3 veces, de 1 en 1000 personas a 1 en 250 personas. En los
estudios de casos y controles, las pacientes que presentan TVP/PE con
anticonceptivos orales o durante el embarazo a menudo tienen esta mutación. La
homocigosidad en este locus (herencia de dos genes defectuosos) aumenta el
riesgo de DVT/PE de 30 a 80 veces, a 1 de cada 12 personas.
4. Gen de la protrombina: la mutación G20210A del gen
de la protrombina se produce en la región 3' no traducida del gen en lugar de
en la secuencia codificante. Estabiliza los niveles de ARNm de protrombina y,
por lo tanto, aumenta el nivel de protrombina en el plasma en estado
estacionario entre un 25 % y un 30 %. Esto da como resultado una mayor
generación de trombina. El curso clínico es bastante similar al factor V
Leiden. Es un segundo ejemplo de una mutación invariable que se ha vuelto común
en la población caucásica. De nuevo, se desconoce la posible ventaja de portar
esta mutación y mantenerla en la población. Hay algunos informes de que los
pacientes con la mutación del gen de la protrombina pueden tener una mayor
incidencia de EP que aquellos con el defecto del factor V de Leiden.
Anticuerpo antifosfolípido
El síndrome de anticuerpos antifosfolípidos, también
llamado anticuerpo anticardiolipina o síndrome anticoagulante lúpico/similar al
lupus, es un trastorno autoinmune que aumenta el riesgo de trombosis arterial y
venosa del paciente (Tabla 3).

Tabla 3. Relación entre estado Trombofílico y Riesgo
de Tromboembolismo Venoso.(OCP anticonceptivos orales)
El mecanismo de inducción de un estado de
hipercoagulabilidad sigue siendo especulativo y muchos pacientes tienen el
trastorno pero permanecen asintomáticos. Las dos pruebas más solicitadas son la
medición de anticuerpos anticardiolipina y la detección de un inhibidor similar
al lupus. Si la prueba de detección es positiva, se realiza una prueba de
confirmación utilizando fosfolípidos en fase hexagonal. También se encuentran
disponibles pruebas de anticuerpos anti-B2GPI y antiprotrombina. Existe alguna
evidencia de que los pacientes con anticuerpos anticardiolipina que también
reaccionan con B2GPI son más propensos a la trombosis. La serología es
complicada, aunque la mayoría de los pacientes, pero no todos, son positivos
tanto en la prueba de anticoagulantes lúpicos como en la de anticardiolipina.
Una vez que un paciente tiene un evento trombótico
inicial, el riesgo de recurrencia es lo suficientemente alto como para que los
pacientes generalmente reciban anticoagulación indefinida o de por vida. La
mayoría de los pacientes tienen TVP o EP, un accidente cerebrovascular o un
evento arterial coronario. Los pacientes raros pueden desarrollar un trastorno
más agresivo; el catastrófico síndrome de anticuerpos antifosfolípidos, con
trombosis potencialmente mortal en múltiples sitios. Se recomienda la
plasmaféresis, además de la anticoagulación vigorosa, para estos pacientes.
Aunque la terapia estándar para un evento trombótico
es heparina seguida de mantenimiento con un anticoagulante de warfarina, existe
alguna evidencia de que un curso del anticuerpo anti-CD20 rituximab puede
reducir o eliminar los anticuerpos anticardiolipina y reducir el riesgo de
tromboembolismo. En algunas series pequeñas publicadas, aproximadamente el 50%
de los pacientes tratados respondieron y pudieron suspender la terapia
anticoagulante.
FUENTE:
The
Brigham Intensive Review of
Internal Medicine. (2022)
Ajay K. Singh, MBBS, FRCP, MBA
Joseph Loscalzo, MD, PhD
No hay comentarios:
Publicar un comentario