La glándula suprarrenal está formada por la corteza y
la médula. La corteza suprarrenal secreta tres clases de hormonas esteroides:
glucocorticoides, mineralocorticoides y andrógenos. La zona glomerulosa externa
secreta el mineralocorticoide aldosterona, que desempeña un papel clave en el
mantenimiento del volumen intravascular y la homeostasis del potasio. La zona
fasciculada central produce cortisol, un glucocorticoide y mineralocorticoide,
que es un actor crucial en la respuesta al estrés y un factor importante en el
metabolismo y las funciones inmunitarias. La capa interna, la zona reticularis,
produce andrógenos, que sirven como precursores de testosterona y
androstenediona; juegan un papel en el desarrollo de las características sexuales
secundarias en las mujeres.
GLUCOCORTICOIDES
La corteza suprarrenal, al ser estimulada por la
hormona adrenocorticotrópica (ACTH) a través de una proteína reguladora aguda
esteroidogénica (StAR), capta el colesterol, el sustrato principal para la
esteroidogénesis. Las hormonas específicas para las tres zonas de la corteza
suprarrenal se producen luego a través de una serie de pasos coordinados de las
enzimas del citocromo P450. El cortisol circula en el plasma como cortisol
libre y unido a proteínas y metabolitos de cortisol. El cortisol es el
principal glucocorticoide endógeno fisiológico que actúa uniéndose a los
receptores de glucocorticoides intranucleares que se expresan en muchos
tejidos. El cortisol también es un potente agonista de los receptores de
mineralocorticoides capaz de inducir efectos similares a los de la aldosterona;
sin embargo, la coexpresión de la enzima 11-beta-hidroxiesteroide
deshidrogenasa (11β-HSD II) con el receptor de mineralocorticoides en el riñón
asegura la inactivación de la mayor parte del cortisol a cortisona y, por lo
tanto, previene un estado de exceso de mineralocorticoides. El cortisol libre o
no unido, que es aproximadamente el 5% del cortisol total, es la hormona
fisiológicamente activa que actúa en los tejidos.
REGULACIÓN DEL EJE HIPOTÁLAMO-PITUITARIO-ADRENAL
La ACTH secretada por la hipófisis anterior regula la
síntesis de cortisol suprarrenal. La ACTH se procesa a partir de una molécula
precursora grande, la proopiomelanocortina (POMC), junto con otros péptidos,
como la beta lipotropina, las endorfinas y la hormona estimulante de los
melanocitos. La hormona liberadora de corticotropina (CRH), producida en el
hipotálamo, estimula la liberación de ACTH y sus péptidos relacionados (fig.
50.1).

Figura 50.1. Regulación de retroalimentación del
sistema hipotálamo-pituitario-suprarrenal (HPA). El eje HPA. Los sitios
principales para el control de retroalimentación del cortisol plasmático son la
glándula pituitaria, el hipotálamo y los centros superiores del cerebro. Hay un
ciclo de retroalimentación corto que implica la inhibición de la hormona
liberadora de corticotropina (CRH) por la hormona adrenocorticotrópica (ACTH).
Hay un control de retroalimentación negativa del cortisol sobre la hipófisis y
el hipotálamo.
Las citocinas inflamatorias en respuesta al estrés
conducen a un aumento del cortisol a través del hipotálamo y el cortisol
producido, a su vez, suprime las citocinas proinflamatorias. (–) Supresión; (+)
estimulación; DHEA, dehidroepiandrostenediona; IL, interleucina; POMC,
proopiomelanocortina; TNF, factor de necrosis tumoral. (De Trikudanathan S,
Dluhy RG. Disfunción cortical suprarrenal. En: Blake MA, Boland G, eds. Adrenal
Imaging. Nueva York: Humana Press; 2009:35–56.)
Varios factores influyen en la liberación de ACTH:
CRH, arginina vasopresina (AVP), ritmo circadiano, estrés y niveles de cortisol
libre. La ACTH tiene un patrón de secreción pulsátil y sigue un ritmo
circadiano, con niveles máximos antes de despertar y valores mínimos al final
de la noche. El patrón de sueño-vigilia, que se ve alterado por los viajes de
larga distancia a través de zonas horarias o por el trabajo nocturno, tarda
unas 2 semanas en restablecerse. El estrés como la fiebre, la cirugía, la
hipoglucemia, el ejercicio y las emociones agudas desencadenan la liberación de
CRH, AVP y, posteriormente, ACTH; también se activa el sistema nervioso
simpático. La interacción inmunoendocrina ocurre cuando las citocinas
proinflamatorias (en particular, la interleucina 1, la interleucina 6 y el
factor de necrosis tumoral α) aumentan los efectos de la CRH y la AVP sobre la
secreción de ACTH. Finalmente, el cortisol plasmático libre ejerce un control
de retroalimentación negativa de la secreción de ACTH, por lo que el cortisol
inhibe la transcripción del gen POMC en la hipófisis anterior y la secreción de
CRH y AVP en el hipotálamo. El cortisol también estimula los centros cerebrales
superiores (como el hipocampo y el sistema reticular) e inhibe el locus
coeruleus/sistema simpático. La administración crónica de corticosteroides
suprime el eje hipotálamo-pituitario-suprarrenal (HPA), que persiste durante
meses después de la interrupción del tratamiento.
Acciones fisiológicas y fisiopatológicas de los
glucocorticoides.
Los glucocorticoides juegan un papel fundamental en el
metabolismo intermedio de carbohidratos, proteínas y grasas. Los
glucocorticoides aumentan la concentración de glucosa en sangre aumentando la
síntesis de glucógeno hepático y estimulando la gluconeogénesis. Los
glucocorticoides también ejercen una acción antiinsulina en los tejidos
periféricos al reducir la captación de glucosa. En consecuencia, el aumento de
las acciones de los glucocorticoides produce resistencia a la insulina y un
aumento de las concentraciones de glucosa en sangre en el marco de un aumento
del catabolismo de proteínas y lípidos.
El exceso de cortisol conduce a un aumento del
depósito de tejido adiposo en el centro de las vísceras en oposición a la
periferia. El exceso de glucocorticoides causa sarcopenia por acciones
catabólicas, así como por la reducción de la síntesis de proteínas en el
músculo. En el esqueleto, la actividad osteoblástica se inhibe dando lugar a
osteoporosis en exceso de glucocorticoides. Los glucocorticoides suprimen las
citocinas inflamatorias y alteran la inmunidad mediada por células. Los
glucocorticoides aumentan el recuento de neutrófilos por la desmarginación de
los neutrófilos con el agotamiento de los eosinófilos. Los cambios en los
niveles de cortisol afectan el estado de ánimo y el sueño, lo que implica que
el cerebro es un objetivo importante de esta hormona. El hipercortisolismo está
fuertemente asociado con el estado de ánimo deprimido, la depresión y el
insomnio.
Síndrome de Cushing
El síndrome de Cushing resulta de la exposición
prolongada e inapropiada a niveles elevados de glucocorticoides. El
hipercortisolismo endógeno puede ser causado por la secreción excesiva de ACTH
de la hipófisis (enfermedad de Cushing 70 %) o por tumores neuroendocrinos no
hipofisarios ectópicos (15 %) o por la secreción excesiva de cortisol por
tumores suprarrenales (15 %). Sin embargo, la causa más frecuente del síndrome
de Cushing es la iatrogenia por prescripción médica de glucocorticoides.
El cuadro 50.1 enumera las causas del síndrome de
Cushing.

Cuadro 50.1 Causas del síndrome de Cushing.
Las causas raras del síndrome de Cushing independiente
de ACTH incluyen hiperplasia macronodular y enfermedad adrenocortical nodular
pigmentada primaria (PPNAD, hiperplasia micronodular). PPNAD puede ser
esporádica o, más a menudo, una parte del complejo de Carney. El complejo de
Carney es un trastorno autosómico dominante caracterizado por pigmentación de
la piel, tumores endocrinos (principalmente PPNAD) y tumores no endocrinos como
mixomas cutáneos, mixomas cardíacos y schwannomas.
Características clínicas del síndrome de Cushing
Las características prominentes del síndrome de
Cushing incluyen obesidad central o troncal con depósitos de grasa aumentados
en sitios distintivos como el área dorsocervical (giba de búfalo), las
almohadillas de grasa supraclaviculares y el lecho mesentérico. Las
extremidades carecen de grasa y parecen delgadas. Los pacientes pueden
desarrollar cara de luna llena, hirsutismo y plétora facial. Los signos de
pérdida de proteínas se observan de manera característica con piel delgada,
fácil formación de hematomas, estrías cutáneas violáceas anchas y miopatía
proximal. La osteoporosis puede ocurrir con fracturas vertebrales. La
intolerancia a la glucosa ocurre debido a la resistencia a la insulina, con
diabetes mellitus manifiesta en alrededor del 20% de los pacientes. Los
estudios de imagen muestran esteatosis hepática y aumento de la grasa visceral.
El exceso de cortisol predispone a la hipertensión y por lo tanto aumenta el
riesgo cardiovascular. En las mujeres, el aumento de los niveles de andrógenos
suprarrenales (cuando el síndrome de Cushing está mediado por ACTH) puede
provocar acné, hirsutismo y anomalías menstruales, como oligomenorrea y
amenorrea. Pueden producirse disfunciones emocionales que van desde la
irritabilidad hasta la depresión o incluso una psicosis franca. Las infecciones
de heridas son comunes y contribuyen a la mala cicatrización de heridas. El
espectro de presentación clínica es amplio y se superpone con muchas afecciones
comunes, como la obesidad simple, y por lo tanto, el diagnóstico puede ser un
desafío. La morbilidad y la mortalidad del síndrome de Cushing se deben
principalmente a complicaciones cardiovasculares, incluida la trombosis,
seguida de causas infecciosas.
Detección del síndrome de Cushing
Las pruebas iniciales para el síndrome de Cushing
deben realizarse en pacientes con características clínicas que sugieran el
síndrome de Cushing o en pacientes con tumores suprarrenales descubiertos
incidentalmente. Es razonable evaluar a los pacientes con características
inusuales para su edad que podrían reflejar hipercortisolismo, como
osteoporosis, hipertensión o hematomas fáciles. Los estudios han demostrado que
el síndrome de Cushing prevalece en el 2% al 5% de los pacientes diabéticos mal
controlados.
La detección inicial del síndrome de Cushing debe
demostrar un aumento de la producción de cortisol y/o la falta de supresión de
la secreción de cortisol cuando se administra un glucocorticoide exógeno
(dexametasona). Una vez que se determina el diagnóstico de hipercortisolismo,
se debe buscar la etiología. Las siguientes pruebas son útiles y
complementarias como estudios diagnósticos iniciales para el síndrome de
Cushing: medición de los niveles de cortisol libre urinario (CLU), cortisol
salival nocturno y prueba de supresión de dexametasona en dosis bajas (DST) (dexametasone
supression test), durante la noche.
La medición de CLU de 24 horas (junto con la
creatinina urinaria para determinar la integridad de la recolección) es útil
para diagnosticar el hipercortisolismo. Mide el cortisol que no se une a la
globulina transportadora de corticosteroides, que es filtrado por el riñón sin
cambios y no se reabsorbe. El CLU no debe medirse en pacientes con
insuficiencia renal de moderada a grave. El CLU también puede ser normal si un
paciente tiene una enfermedad cíclica o un síndrome de Cushing leve. Se
observan resultados positivos falsos en cualquier estado fisiológico que
aumente la producción de cortisol; por lo tanto, pueden ser necesarias dos
mediciones realizadas en ocasiones separadas.
La secreción de cortisol sigue un patrón diurno con
niveles máximos en la mañana y nadir en la noche. El cortisol normalmente
alcanza un nadir por la noche; sin embargo, este ritmo circadiano normal se
pierde en el síndrome de Cushing. Por lo tanto, la detección de un cortisol
elevado a altas horas de la noche o de medianoche es otro método sensible para
detectar el hipercortisolismo. Un cortisol salival de medianoche, obtenido a
través de hisopado de la mucosa oral, es la prueba más utilizada para esta
indicación.
Una prueba de detección ambulatoria simple es el DST
nocturno (1 mg de dexametasona entre las 10 p. m. y la medianoche, seguido de
una evaluación de cortisol matutino a las 8 a 9 a. m.). Un nivel de cortisol
matutino posterior a la dexametasona >5 μg/dL se considera anormal, y un
nivel >1.8 μg/dL se considera sugestivo de hipercortisolismo endógeno.
Investigaciones para identificar la causa del síndrome
de Cushing
Una vez que se confirma el diagnóstico de
hipercortisolismo, el siguiente paso es determinar si la etiología es ACTH
dependiente o ACTH independiente. Un nivel de ACTH bajo o indetectable (<9
pg/ml) confirma el diagnóstico de trastornos suprarrenales primarios. También
hay supresión en el sulfato de dehidroepiandrosterona plasmática (DHEAS) porque
la producción de andrógenos suprarrenales se reduce como resultado de la
supresión de ACTH. En los carcinomas suprarrenales, el hipercortisolismo suele
ir acompañado de un aumento de la secreción de andrógenos. La producción de
esteroides en el carcinoma suprarrenal suele ser resistente a la estimulación
con ACTH y la supresión con dexametasona. Los pacientes con trastornos
suprarrenales primarios deben someterse a una tomografía computarizada de alta
resolución del abdomen. En los microadenomas hipofisarios secretores de ACTH
(enfermedad de Cushing), las concentraciones de ACTH son inapropiadamente
normales o moderadamente elevadas (27 a 136 pg/mL), mientras que en los
macroadenomas hipofisarios y el síndrome de ACTH ectópica, las concentraciones
de ACTH pueden aumentar dos o tres veces.
Para distinguir las etiologías del síndrome de Cushing
dependiente de ACTH, se pueden usar pruebas de supresión con dosis altas de
dexametasona (prueba nocturna de 8 mg o 2 mg cada 6 horas durante 2 días). El
macroadenoma hipofisario y la producción ectópica de ACTH a menudo no muestran
supresión, mientras que suele haber cierta supresión de ACTH y cortisol en los
microadenomas hipofisarios secretores de ACTH. Este método funcional para
localizar la fuente del exceso de ACTH no es muy confiable y, por lo tanto,
puede ser necesario el uso de imágenes en combinación con el muestreo del seno
petroso inferior (IPSS).
La resonancia magnética para evaluar una masa
hipofisaria es el estudio de imagen inicial cuando se sospecha síndrome de
Cushing dependiente de ACTH; sin embargo, es posible que no siempre demuestre
una lesión hipofisaria en pacientes con enfermedad de Cushing. Algunos adenomas
secretores de ACTH son demasiado pequeños para ser vistos de manera confiable
en la resonancia magnética. Tenga en cuenta que del 10% al 20% de los pacientes
normales tienen “incidentalomas” hipofisarios que no funcionan. En la mayoría
de las circunstancias, se necesita IPSS para probar la hipersecreción
hipofisaria de ACTH. La sangre de cada mitad de la pituitaria drena en el seno
cavernoso y luego en el seno petroso inferior ipsolateral. El cateterismo y el
muestreo venoso para la medición de ACTH de ambos senos petrosos
simultáneamente en comparación con una muestra periférica diferenciarían una
fuente hipofisaria de una fuente ectópica. En el tumor hipofisario secretor de
ACTH, la relación entre las concentraciones de ACTH del seno petroso inferior y
la sangre periférica extraída simultáneamente sería mayor de 2 veces basalmente
y mayor de 3 veces después de la inyección de CRH. Por lo tanto, IPSS es una
prueba altamente sensible y específica para distinguir entre fuentes
hipofisarias y no hipofisarias de exceso de ACTH. Sin embargo, IPSS es
técnicamente exigente y pueden ocurrir complicaciones como trombosis; por lo
tanto, esta prueba debe realizarse en un centro experimentado.
Para localizar las fuentes de producción ectópica de
ACTH, es razonable comenzar con imágenes del tórax y el abdomen para buscar una
fuente de ACTH pulmonar o pancreática. La exploración con octreotida también
puede ser útil para obtener imágenes de tumores neuroendocrinos productores de
ACTH, como los carcinoides.
Diagnóstico diferencial
Síndrome de pseudo-Cushing
La obesidad, el alcoholismo crónico y la depresión
pueden imitar las anomalías bioquímicas del hipercortisolismo. Por ejemplo, la
ingesta crónica y excesiva de alcohol y la depresión pueden causar elevaciones
leves del CLU, disminución del ritmo circadiano y resistencia a la supresión
con dexametasona.
Sin embargo, estos pacientes generalmente no tienen
los signos y síntomas clínicos más confiables del síndrome de Cushing, como
miopatía proximal y fácil formación de hematomas. Después de la interrupción
del alcohol o con el alivio de la depresión, las pruebas de esteroides vuelven
a la normalidad. Cuando esté disponible (típicamente en centros médicos
académicos seleccionados), se puede realizar una DST estimulada con CRH para
diferenciar el síndrome de pseudo-Cushing del hipercortisolismo endógeno y
autónomo verdadero.
Manejo
La resección quirúrgica del adenoma hipofisario
mediante el abordaje transesfenoidal es la primera línea de tratamiento para la
enfermedad de Cushing (tumor hipofisario secretor de ACTH). La remisión en
manos de un cirujano experimentado está en el rango de 65% a 90% para
microadenomas y 50% para macroadenomas. Después de extirpar el adenoma
hipofisario productor de ACTH, se suprimen los corticotropos normales; por lo
tanto, los pacientes suelen necesitar tratamiento con glucocorticoides en el
posoperatorio hasta que se recupera el eje HPA. Un nivel de cortisol sérico
matutino posoperatorio de <2 μg/dL el día después de la cirugía sugiere
remisión y posible curación quirúrgica. En el pasado, la suprarrenalectomía
bilateral se consideró como una opción para la enfermedad de Cushing; sin
embargo, la extirpación de las glándulas suprarrenales puede llevar al
desarrollo del síndrome de Nelson en 10% a 20% de los pacientes (un
macroadenoma hipofisario secretor de ACTH agresivo). Se cree que el síndrome de
Nelson es la consecuencia de la eliminación de la fuente de retroalimentación
negativa sobre la ACTH, lo que da como resultado un marcado crecimiento y
función del adenoma secretor de ACTH. La irradiación hipofisaria se puede utilizar
para
pacientes con recurrencia postoperatoria y en síndrome
de Nelson. En otros centros, se han utilizado técnicas de bisturí gamma y
estereotácticas para tratar los adenomas hipofisarios.
En el síndrome de ACTH ectópica, la terapia dirigida
al tumor que implica la resección del tumor primario (p. ej., carcinoide
bronquial) puede conducir a la curación. Sin embargo, el pronóstico sigue
siendo malo para los tumores pulmonares de células pequeñas y la terapia médica
que inhibe la esteroidogénesis está indicada para los síntomas de exceso de
cortisol.
Se prefiere la suprarrenalectomía laparoscópica o
retroperitoneoscópica para los adenomas suprarrenales. Los carcinomas
suprarrenales tienen un mal pronóstico con tasas de supervivencia a 5 años
deprimentes. Por lo general, los carcinomas suprarrenales no son radiosensibles
y responden mal a las quimioterapias sistémicas, aunque se ha demostrado que el
mitotano mejora la supervivencia libre de enfermedad si se administra de forma
adjunta después de la resección quirúrgica de la neoplasia. El mejor predictor
del resultado es la capacidad de lograr una resección quirúrgica completa.
Terapias médicas para el síndrome de Cushing
Se pueden usar fármacos para tratar el
hipercortisolismo mediante la inhibición de la esteroidogénesis: metirapona,
ketoconazol y mitotano. La metirapona inhibe la 11β hidroxilasa, mientras que
el ketoconazol bloquea múltiples enzimas dependientes del citocromo P450
esteroidogénico suprarrenal. Estos fármacos se pueden utilizar antes de la operación
o como tratamiento complementario después de la cirugía o la radioterapia. El
mitotano inhibe la esteroidogénesis, pero en algunos pacientes también es
citotóxico para la glándula suprarrenal. Su uso es principalmente para el
carcinoma suprarrenal debido a su citotoxicidad.
Pasireotide, un nuevo análogo de somatostatina con
alta afinidad por el subtipo 5 del receptor de somatostatina, ha sido aprobado
recientemente por la Administración de Drogas y Alimentos de EE. UU. para la
enfermedad de Cushing. Debido a que los adenomas productores de ACTH expresan
mucho el subtipo 5 del receptor de somatostatina, la activación de este
receptor inhibe la secreción de ACTH. En ensayos clínicos, la pasireotida
redujo la UFC y mejoró las características clínicas del hipercortisolismo. Cabe
destacar que se observó una alta frecuencia de hiperglucemia.
Insuficiencia suprarrenal
La insuficiencia suprarrenal primaria (enfermedad de
Addison) resulta de la destrucción de la corteza suprarrenal, lo que resulta en
una deficiencia en la producción de aldosterona, cortisol y andrógenos
suprarrenales. La insuficiencia suprarrenal secundaria es la consecuencia de la
disminución de la producción de ACTH que conduce a una reducción de la
secreción de cortisol y andrógenos suprarrenales; la producción de aldosterona
es normal porque el eje renina-angiotensina permanece intacto en tales
pacientes. Aunque la enfermedad de Addison es poco común, conlleva una
morbilidad y mortalidad significativas si no se trata.
Etiología
La causa más común de la enfermedad de Addison es la
adrenalitis autoinmune, y la mayoría de los pacientes tienen autoanticuerpos
dirigidos contra la 21 hidroxilasa y las enzimas de escisión de la cadena
lateral. La insuficiencia suprarrenal primaria puede ocurrir como parte de los
síndromes poliendocrinos autoinmunes (SAF) І y ІІ.
En el mundo en desarrollo, la insuficiencia
suprarrenal primaria a menudo es causada por infecciones, especialmente
tuberculosis. Sin embargo, cualquier proceso infiltrativo (infección, malignidad,
hemorragia) que afecte ambas cortezas suprarrenales puede resultar en
insuficiencia suprarrenal primaria. Debido a que las terapias contra el cáncer
permiten que los pacientes con neoplasias malignas metastásicas sobrevivan más
tiempo, y debido a que a los pacientes con trasplantes de órganos con
inmunosupresión les va mejor, la prevalencia de insuficiencia suprarrenal
primaria atribuida a neoplasias malignas invasivas e infecciones fúngicas está
aumentando. La insuficiencia suprarrenal secundaria puede ser causada por
cualquier proceso que altere el hipotálamo y/o la hipófisis, incluidos tumores
malignos, infecciones, inflamación o hemorragia. La causa más común de
insuficiencia suprarrenal secundaria es probablemente la medicación iatrogénica
inducida por glucocorticoides. El uso de glucocorticoides exógenos puede
suprimir la secreción central de ACTH al unirse al receptor de glucocorticoides
hipotalámico e hipofisario y, por lo tanto, provocar una disminución de la
secreción suprarrenal de cortisol y, si se prolonga, atrofia de la corteza
suprarrenal. Los opioides de acción corta también pueden suprimir
transitoriamente la secreción de ACTH. Debido a que los opioides endógenos son
secretados por el hipotálamo y la hipófisis (son un producto de la escisión de
POMC, al igual que la ACTH), los receptores de opioides se expresan en el
hipotálamo y la hipófisis. Aunque faltan estudios de cohortes grandes, una
colección de observaciones anecdóticas sugiere que los opioides, especialmente
los opioides de acción corta que se unen al receptor mu, reducen
transitoriamente la ACTH y el cortisol.
No se ha estudiado bien si estos pueden dar lugar a un
síndrome clínico de insuficiencia suprarrenal o no, pero debe tenerse en cuenta
al evaluar a pacientes con características preocupantes para la insuficiencia
suprarrenal que también reciben terapia con opioides. En el cuadro 50.2 se
enumeran otras causas de insuficiencia suprarrenal.

Box 50.2. 2 causas de la insuficiencia suprarrenal
Características clínicas
Los síntomas de la insuficiencia suprarrenal crónica
no son específicos e incluyen fatiga, debilidad, apatía, anorexia y pérdida de
peso. En ocasiones, los síntomas gastrointestinales como náuseas, vómitos,
diarrea y calambres abdominales pueden ser el único síntoma de presentación. Un
signo específico de insuficiencia suprarrenal primaria es la hiperpigmentación
cutánea y de la membrana mucosa, que ocurre debido a la hormona estimulante de
los melanocitos y la ACTH elevadas por la ausencia de retroalimentación
negativa de cortisol en el hipotálamo y la hipófisis. El oscurecimiento de la
piel se observa típicamente en las áreas expuestas al sol, cicatrices
recientes, pliegues palmares y mucosa bucal y vaginal. La hipotensión
ortostática puede ser marcada en la insuficiencia suprarrenal primaria debido a
la deficiencia de aldosterona; el ansia de sal es una queja frecuente. Las
mujeres pueden notar pérdida de vello púbico y axilar, como resultado de la
deficiencia de andrógenos suprarrenales. Las anomalías bioquímicas incluyen
hiponatremia (frecuente), hiperpotasemia, hipoglucemia, elevación de la urea en
sangre, hipercalcemia leve, anemia normocítica leve, linfocitosis y
eosinofilia. En la insuficiencia suprarrenal primaria, la hiponatremia ocurre
debido a la deficiencia de aldosterona y la pérdida de sodio, mientras que en
el hiposuprarrenalismo secundario, es dilucional debido a la deficiencia de
cortisol, que se asocia con niveles elevados de hormona antidiurética y
eliminación ineficaz de agua libre.
La insuficiencia suprarrenal aguda, cuando es causada
por una hemorragia suprarrenal o precipitada por una infección aguda, se
presenta como hipotensión, insuficiencia circulatoria aguda, confusión, dolor
abdominal y fiebre; el reconocimiento rápido es extremadamente importante.
En la insuficiencia suprarrenal secundaria, la
palidez, la cefalea, el vello axilar y púbico escaso y los síntomas visuales
pueden indicar una enfermedad hipotalámica-hipofisaria. No se observa
hiperpotasemia porque hay una secreción normal de aldosterona.
Los pacientes con insuficiencia suprarrenal secundaria
generalmente no presentan hipotensión ni complicaciones hemodinámicas porque se
mantiene la regulación de la aldosterona y, por lo tanto, el volumen
intravascular es generalmente normal. Sin embargo, en situaciones de estrés
(como infección o traumatismo), la deficiencia relativa de cortisol puede dar
lugar a un empeoramiento progresivo del síndrome que puede simular la
fisiopatología hemodinámica observada en la insuficiencia suprarrenal primaria.
Diagnóstico
Un nivel matutino de cortisol plasmático de ≤3 μg/dL
es diagnóstico de insuficiencia suprarrenal manifiesta y excluye la necesidad
de realizar más pruebas; niveles ≥18 μg/dL descartan el trastorno. Los niveles
matutinos de cortisol que son >10 a 15 μg/dl suelen reflejar un eje HPA
normal cuando la probabilidad previa a la prueba de insuficiencia suprarrenal
es relativamente baja. No existe un valor de corte absoluto para un cortisol
matutino normal o anormal cuando los niveles son >5 μg/dL; más bien, se debe
usar un juicio clínico cuidadoso para determinar si el cortisol matutino máximo
que se evalúa es "apropiado" para el escenario clínico. Esto puede
dificultar el diagnóstico de casos sutiles de insuficiencia suprarrenal.
La prueba diagnóstica más utilizada para la
insuficiencia suprarrenal es la prueba de estimulación con ACTH, en la que se
administran 250 μg de cosintropina por vía intramuscular o intravenosa y se
mide la respuesta del cortisol a los 0, 30 y 60 minutos. La respuesta normal es
una respuesta de cortisol basal o máxima >18 μg/dl. Esta prueba es útil para
diagnosticar la destrucción primaria del tejido y la insuficiencia suprarrenal
secundaria prolongada. Esta prueba puede ser normal en pacientes con
insuficiencia suprarrenal secundaria leve o de aparición reciente. En plasma
temprano en la mañana, el nivel de ACTH es útil para distinguir la
insuficiencia suprarrenal primaria de la secundaria si los niveles de cortisol
son anormales. Los valores plasmáticos de ACTH suelen estar elevados (>100
pg/mL) en la insuficiencia suprarrenal primaria a diferencia del
hiposuprarrenalismo secundario, donde los valores plasmáticos de ACTH pueden
ser bajos o “inapropiadamente” normales. Otras pruebas, como la prueba de
tolerancia a la insulina, la prueba de metirapona y la prueba de CRH, se
utilizan con poca frecuencia para diagnosticar la insuficiencia suprarrenal
secundaria.
La tomografía computarizada de las glándulas
suprarrenales puede mostrar agrandamiento (p. ej., hemorragia) o calcificación,
según la etiología de la insuficiencia suprarrenal. En la insuficiencia
suprarrenal secundaria, hay una secreción normal de aldosterona y no se observa
hiperpotasemia. Por lo general, en estos pacientes se necesitan resonancias
magnéticas hipofisarias y la evaluación de las funciones de la hipófisis
anterior debido a las deficiencias concomitantes de otras hormonas
hipofisarias.
Las personas que reciben terapia con esteroides en
dosis altas a largo plazo desarrollarán una supresión prolongada de HPA que
conducirá a la atrofia suprarrenal. La recuperación puede tardar meses o años
después de la suspensión de los glucocorticoides. Los niveles de cortisol
temprano en la mañana y las pruebas de estimulación con ACTH deben usarse para
evaluar la recuperación suprarrenal.
Diagnóstico diferencial
Síntomas crónicos inespecíficos como fatiga, debilidad
y malestar general deben hacer posible el diagnóstico de insuficiencia
suprarrenal. Cuando tiene un inicio insidioso, la insuficiencia suprarrenal se
confunde con frecuencia con el síndrome de fatiga crónica. Ocasionalmente,
estos pacientes han sido mal diagnosticados con anorexia nerviosa o depresión.
Sin embargo, la hiperpigmentación, la pérdida de peso y los síntomas
gastrointestinales deben alertar al médico para considerar la insuficiencia
suprarrenal. También es razonable buscar otras enfermedades autoinmunes
específicas de órganos en el contexto de los síndromes poliglandulares.
Manejo
En el contexto de una crisis suprarrenal, se debe iniciar
inmediatamente el tratamiento parenteral con altas dosis de hidrocortisona
junto con la reposición de líquidos con solución salina normal. En situaciones
no agudas, se deben iniciar dosis de reemplazo de hidrocortisona oral en dosis
de 8 a 10 mg/m2/d en dosis divididas. Para imitar el patrón diurno de secreción
de esteroides, dos tercios de la dosis total se administran por la mañana y un
tercio al final de la tarde con las comidas o los refrigerios. En la
insuficiencia suprarrenal secundaria, solo se necesita terapia con
glucocorticoides.
En la insuficiencia suprarrenal primaria, la
insuficiencia mineralocorticoide se reemplaza con fludrocortisona, administrada
a una dosis diaria de 0,05 a 0,1 mg por vía oral. La actividad de la renina
plasmática, la presión arterial y los electrolitos séricos son parámetros
útiles para titular la dosis de fludrocortisona. En pacientes mujeres, algunos
estudios han sugerido el beneficio del tratamiento con andrógenos con 25 a 50
mg/día de DHEA por vía oral para mejorar la función sexual y el bienestar
general; sin embargo, la respuesta clínica a la DHEA es variable.
La educación del paciente y la terapia de reemplazo
diaria forman la piedra angular en el manejo de la insuficiencia suprarrenal
primaria. Se aconseja a los pacientes que dupliquen la dosis de hidrocortisona
durante los períodos de enfermedad o cirugía intercurrentes. Todos los
pacientes deben usar un brazalete de alerta médica y deben recibir
instrucciones sobre la autoinyección de esteroides si no pueden tomar su dosis
por vía oral.
Regulación del Eje Renina-AngiotensinaAldosterona
La renina se forma en las células yuxtaglomerulares
(JG), ubicadas adyacentes a la arteriola aferente renal del glomérulo. La
renina se estimula cuando disminuye la perfusión renal, como se detecta por la
disminución del suministro distal de iones de cloruro. La renina actúa sobre el
sustrato angiotensinógeno (de origen hepático) para formar angiotensina І. La
angiotensina І se convierte en angiotensina ІІ mediante la enzima convertidora
de angiotensina (ECA). La angiotensina ІІ tiene varias funciones cruciales: es
un potente vasoconstrictor arterial, aumenta directamente la reabsorción de
sodio del túbulo proximal, estimula la liberación de vasopresina (lo que
garantiza que la reabsorción de agua se acompañe de la reabsorción de sodio) y
estimula la zona glomerulosa de la corteza suprarrenal para aumentar la
secreción de aldosterona (fig. 50.2).

Figura 50.2. Sistema renina-angiotensina-aldosterona.
Esta figura muestra la interacción de varias señales de la nefrona en el riñón,
el hígado y la glándula suprarrenal que forman el circuito de retroalimentación
para mantener el volumen de sangre circulante y la secreción de aldosterona.
(–) Supresión; (+) estimulación; ANP, péptido natriurético auricular; DCT,
túbulo contorneado distal; JG, yuxtaglomerular; K+, potasio. (De Trikudanathan
S, Dluhy RG. Disfunción cortical suprarrenal. En: Blake MA, Boland G, eds.
Adrenal Imaging. Nueva York: Humana Press; 2009:35–56.)
El control de la secreción suprarrenal de aldosterona
incluye el sistema renina-angiotensina, potasio y ACTH. La aldosterona cumple
dos funciones importantes: la regulación del volumen de líquido extracelular y
la homeostasis del potasio. La exposición crónica a la aldosterona durante 3 a
5 días conduce a un “escape” de la acción de los mineralocorticoides; después
de un período inicial de retención de sodio y una ganancia de varios
kilogramos, se restablece el equilibrio de sodio. Por lo tanto, no se
desarrolla edema. Un aumento en el péptido natriurético auricular y la
interacción de los factores hemodinámicos renales juegan un papel en el
“escape” de la acción de retención de sodio de la aldosterona. Sin embargo, es
importante darse cuenta de que no hay "escape" de los efectos de
pérdida de potasio de la exposición crónica a los mineralocorticoides.
La acción tóxica no epitelial de la aldosterona
incluye inflamación, necrosis y fibrosis subsiguiente en una variedad de
tejidos, incluidos el corazón, el riñón y la vasculatura. Estas situaciones
fisiopatológicas ocurren cuando los niveles de aldosterona se elevan de manera
inapropiada con una ingesta alta de sal, como en el aldosteronismo primario
(AP). La aldosterona se ha implicado en la fisiopatología de la insuficiencia cardíaca.
Los receptores de mineralocorticoides se sobreexpresan en el tejido cardíaco
defectuoso, lo que lleva a la fibrosis cardíaca. En el estudio RALES
(Randomized Aldactone Evaluation Study), la espironolactona, un antagonista de
los receptores de mineralocorticoides, redujo la mortalidad en pacientes con
insuficiencia cardíaca sistólica en un 30 % y redujo las hospitalizaciones
frecuentes por insuficiencia cardíaca en un 35 %. También se demostró que la
eplerenona, un bloqueador selectivo de los receptores de mineralocorticoides,
reduce la mortalidad y la morbilidad cardiovascular en pacientes con
insuficiencia ventricular izquierda después de un infarto de miocardio (ensayo
EPHESUS [Eplerenone Post–Acute Myocardial Infarction Heart Failure Eficacy and Survival
Study]).
Aldosteronismo primario
Ahora se reconoce que el AP es la forma más común de
hipertensión secundaria, prevalente en casi el 10% de todos los pacientes con
hipertensión. Está causada por la secreción autónoma de aldosterona por un
adenoma suprarrenal unilateral o por una hiperplasia suprarrenal bilateral.
Los pacientes hipopotasémicos con AP presentan
síntomas inespecíficos como calambres musculares, debilidad, dolores de cabeza,
palpitaciones, poliuria y nicturia. La hipertensión normopotasémica sigue
siendo la forma más común de presentación de este trastorno y probablemente
refleja una detección más temprana de la enfermedad. Se debe considerar un
diagnóstico de AP en pacientes hipertensos con hipertensión refractaria, que es
la presión arterial mal controlada con tres agentes antihipertensivos (incluido
un diurético), y en pacientes con hipopotasemia espontánea o inducida por
diuréticos. Además, todos los pacientes con un tumor suprarrenal deben someterse
a pruebas de detección de AP, al igual que los familiares de pacientes con AP o
accidente cerebrovascular hemorrágico antes de los 40 años y los pacientes con
aparición temprana de AP antes de los 25 años. Varios estudios también han
demostrado que los pacientes con AP tienen una mayor morbilidad y mortalidad
cardiovascular en comparación con los pacientes de la misma edad con
hipertensión esencial.
Diagnóstico
La Endocrine Society recomienda el uso de la
proporción de aldosterona:renina (ARR) en plasma para detectar AP. Idealmente,
la prueba debe realizarse por la mañana en un paciente ambulatorio sentado que
ha estado con una ingesta dietética de sal sin restricciones. Aunque ciertos
medicamentos pueden afectar la RRA principalmente al alterar los niveles de
renina (por ejemplo, la espironolactona, la eplerenona, la amilorida, los
inhibidores de la ECA y los bloqueadores de los receptores de angiotensina, y
el triamtereno puede aumentar la renina, mientras que los bloqueadores beta
pueden disminuir la renina), no se recomienda de forma rutinaria que todos de
estos medicamentos se suspenda antes de la detección. El uso de
espironolactona, eplerenona y amilorida son estimulantes particularmente
potentes de la renina y, por lo tanto, las pruebas de ARR cuando se usan estos
medicamentos deben considerarse no interpretables a menos que la renina esté
completamente suprimida o sea indetectable.
El fundamento detrás de la ARR es evaluar el
aldosteronismo independiente de la renina, donde la aldosterona se secreta a
pesar de la relativa ausencia de actividad de la renina plasmática (ARP). Una
ARR >30 (cuando la aldosterona sérica es >15 ng/dl y ARP <1 ng/mL/h)
es altamente sugestiva de AP (fig. 50.3); sin embargo, las concentraciones más
bajas de aldosterona sérica en pacientes con ARR anormal generalmente necesitan
una prueba de confirmación para diagnosticar AP.

La figura 50.3. Algoritmo para el diagnóstico de
aldosteronismo primario. a Control inadecuado de la hipertensión con tres
antihipertensivos (incluido un diurético). b Carga de sal oral/intravenosa: 2 g
de tabletas de cloruro de sodio durante 3 días o 2 L de solución salina
isotónica durante 4 horas por vía intravenosa. Aldosterona urinaria de 24 horas
recolectada el día 3. Examen de TC de protocolo suprarrenal dedicado (ver
texto). APA, adenoma productor de aldosterona; AVS, muestreo venoso
suprarrenal; PA, aldosterona plasmática; PRA, actividad de renina plasmática.
(De Trikudanathan S, Dluhy RG. Disfunción cortical suprarrenal. En: Blake MA,
Boland G, eds. Adrenal Imaging. Nueva York: Humana Press; 2009:35–56.)
Se puede utilizar cualquiera de los siguientes cuatro
procedimientos de confirmación: carga oral de sodio, infusión de solución
salina, supresión con fludrocortisona y provocación con captopril. El criterio
de valoración de estas pruebas es demostrar la autonomía de la secreción de
aldosterona. No hay evidencia adecuada para recomendar una prueba sobre las
demás, y la elección de la prueba a menudo es específica del centro. Para las
pruebas de supresión oral de sodio, se indica a los pacientes que tomen
tabletas de cloruro de sodio (2 g) con cada comida con una dieta normal en sal
durante 4 días. En el cuarto día, una excreción de aldosterona en orina de 24 h
>10 a 12 μg/24 h en presencia de una excreción de sodio en orina >200
mmol/día es diagnóstica de producción autónoma de aldosterona. Cabe señalar que
no se debe realizar una prueba de carga de sodio oral ni una prueba de infusión
de solución salina intravenosa en pacientes con hipopotasemia no corregida,
hipertensión grave no controlada o insuficiencia cardíaca congestiva.
Tras la confirmación bioquímica de AP, los pacientes
candidatos a cirugía y dispuestos a someterse a un procedimiento quirúrgico
pueden proceder a un estudio de localización mediante TAC o RM de
suprarrenales. Los pacientes que no son candidatos quirúrgicos o que no están
dispuestos a someterse a una adrenalectomía potencial pueden tratarse
empíricamente con un antagonista del receptor de mineralocorticoides. El
muestreo venoso suprarrenal (AVS) bilateral se usa a menudo, a pesar de los
hallazgos de imagen, para confirmar la localización y la lateralidad del exceso
de aldosterona. La AVS debe realizarse en un centro con radiólogos
experimentados para garantizar un cateterismo exitoso y minimizar el riesgo de
hemorragia suprarrenal y trombosis venosa.
Tratamiento
A los pacientes con diagnóstico de adenoma productor
de aldosterona (APA) unilateral se les debe ofrecer adrenalectomía
laparoscópica unilateral. Esto puede llevar a mejoras dramáticas tanto en la
presión arterial como en las concentraciones séricas de potasio en todos los
pacientes. La hipertensión se cura en cerca de 50% de los pacientes después de
adrenalectomía unilateral; la hipertensión persistente después de la
adrenalectomía en pacientes con APA es causada por hipertensión esencial
coexistente, edad avanzada, insuficiencia renal y mayor duración de la
hipertensión.
En pacientes con enfermedad suprarrenal bilateral o
cuando los pacientes con AAF no son candidatos quirúrgicos para adrenalectomía,
deben ser tratados con un antagonista de los mineralocorticoides
(espironolactona o eplerenona). En pacientes masculinos que desarrollan efectos
secundarios predecibles relacionados con la dosis de espironolactona, como
ginecomastia, disminución de la libido e impotencia, se debe usar eplerenona,
un antagonista selectivo del receptor de mineralocorticoides desprovisto de
acciones antiandrogénicas y de progesterona. Otros agentes útiles incluyen los
diuréticos ahorradores de potasio amilorida y triamtereno.
Aldosteronismo remediable con glucocorticoides
El aldosteronismo remediable con glucocorticoides
(GRA), heredado como un trastorno autosómico dominante, resulta de una
duplicación de genes quiméricos, que es el resultado de un cruce desigual entre
los genes homólogos de 11β-hidroxilasa y aldosterona sintasa. Como resultado,
hay expresión ectópica de la enzima aldosterona sintasa en la zona fasciculada
productora de cortisol, bajo la regulación de la ACTH. GRA se caracteriza por
hipertensión de inicio temprano, accidente cerebrovascular hemorrágico y
niveles de renina plasmática suprimidos. Las pruebas genéticas mediante la
técnica de transferencia de Southern deben considerarse para pacientes con PA
con antecedentes familiares de PA o antecedentes familiares de accidentes
cerebrovasculares hemorrágicos a una edad temprana (<30 años) o con
hipertensión de inicio temprano. Una recolección de orina de 24 horas revelaría
una marcada elevación en los niveles de los esteroides “híbridos”
18-oxocortisol y 18-OH-cortisol. El tratamiento con un glucocorticoide de
acción prolongada suprimirá la secreción de aldosterona regulada por ACTH. Se
debe usar la dosis efectiva más pequeña para controlar la presión arterial y
minimizar el riesgo de síndrome de Cushing. Los tratamientos alternativos
incluyen el receptor de mineralocorticoides y los antagonistas de los canales
epiteliales de sodio.
Hiperaldosteronismo secundario
En el hiperaldosteronismo secundario, hay un aumento
apropiado en la producción de aldosterona causado por niveles circulantes
elevados de renina. La producción elevada de renina puede ocurrir en el
contexto de un volumen sanguíneo circulante efectivo reducido (p. ej.,
cirrosis, síndrome nefrótico e insuficiencia cardíaca congestiva) o una
perfusión renal disminuida. La estenosis aterosclerótica de la arteria renal o
las hiperplasias fibromusculares son ejemplos de sobreproducción de renina
causada por la disminución de la perfusión renal. Estos pacientes pueden tener
alcalosis hipopotasémica como resultado del hiperaldosteronismo y aumentos de
moderados a marcados en la actividad de la renina plasmática.
Otras causas de hipermineralocorticoidismo
Hipoaldosteronismo con actividad de renina plasmática
suprimida
El exceso aparente de mineralocorticoides (AME) puede
ocurrir tanto en formas hereditarias como adquiridas de alteración de la
actividad de la enzima renal 11β-HSD II. La deficiencia enzimática da como
resultado la incapacidad de degradar el cortisol a la cortisona biológicamente
inactiva en los túbulos renales. Como resultado, el cortisol se une al MR
ejerciendo acciones mineralocorticoides. La forma adquirida de este síndrome
está causada por la ingestión de ciertos regaliz o tabaco de mascar, que
contiene ácido glicirrícinico. Estos pacientes muestran hipertensión e
hipopotasemia; Se suprimen los niveles de PRA y aldosterona. Los niveles de
cortisol son normales porque el circuito de retroalimentación de ACTH está
intacto. Se pueden usar pequeñas dosis de dexametasona para suprimir la
producción endógena de cortisol.
El síndrome de Liddle es un trastorno autosómico
dominante causado por mutaciones de ganancia de función en las subunidades del
canal epitelial renal de sodio que normalmente está regulado por la
aldosterona. La activación constitutiva del canal da como resultado retención
de sodio, hipopotasemia y niveles bajos de renina/
niveles de aldosterona.
Hiperaldosteronismo con actividad de renina plasmática
elevada.
Los pacientes con síndrome de Bartter (SB) presentan
alcalosis hipopotasémica, hipercalciuria, presión arterial normal y ausencia de
edema. En el SB, la mutación de pérdida de función en el asa del gen del
cotransportador NaK-2Cl de Henle da como resultado la activación del sistema
renina-angiotensina-aldosterona y, por lo tanto, la pérdida renal de sodio. Los
pacientes con síndrome de Gitelman (SG) tienen características similares al SB
excepto que son hipocalciúricos. La GS se debe a mutaciones de pérdida de
función en el cotransportador de Na-Cl sensible a las tiazidas en el túbulo
contorneado distal del riñón.
Hipoaldosteronismo
El hipoaldosteronismo hiporreninémico, que suele
ocurrir en adultos diabéticos con insuficiencia renal leve, produce
hiperpotasemia y acidosis metabólica que no guardan proporción con el nivel de
insuficiencia renal (acidosis tubular renal tipo IV). La deficiencia aislada de
aldosterona con niveles bajos de renina también ocurre en el posoperatorio
después de la extirpación de un adenoma productor de aldosterona y después de
un tratamiento prolongado con heparina.
El sistema simpaticoadrenal
El sistema simpático suprarrenal se deriva de la cresta
neural y consta de los ganglios del sistema nervioso simpático y la médula
suprarrenal. La adrenalina, sintetizada en la médula suprarrenal, y la
noradrenalina, en las terminaciones nerviosas periféricas, se forman a partir
del aminoácido tirosina. La enzima limitante de la velocidad en la vía
biosintética es la tirosina hidroxilasa.
El metabolismo de la epinefrina y la norepinefrina a
compuestos biológicamente inactivos por la catecolamina-O-metil transferasa da
como resultado metanefrina y norepinefrina, respectivamente. La oxidación
adicional da como resultado ácido vanililmandélico.
Feocromocitoma y Paraganglioma
Los feocromocitomas son tumores neuroectodérmicos que
surgen de las células cromafínicas. Estos tumores secretores de catecolaminas
surgen principalmente de la médula suprarrenal; si estos tumores de cromafina
surgen en los ganglios parasimpáticos o simpáticos, se denominan
paragangliomas.
Los feocromocitomas pueden ocurrir esporádicamente o
pueden ser parte de un síndrome tumoral hereditario más grande. Estos síndromes
genéticos se reconocen cada vez más y ahora se estima que el 40 % de todos los
feocromocitomas y paragangliomas pueden atribuirse a una mutación conocida de
la línea germinal con implicaciones no solo para el paciente sino también para
su familia. La presentación clínica de los tumores productores de catecolaminas
varía desde la hipertensión esencial hasta las clásicas crisis hipertensivas
paroxísticas. La falta de diagnóstico y tratamiento del feocromocitoma puede
provocar crisis hipertensivas y la muerte.
Características clínicas
Clásicamente, los pacientes se presentan con
paroxismos de hipertensión grave y palpitaciones; sin embargo, la hipertensión
sostenida ocurre en aproximadamente el 50% de los pacientes. La sudoración
generalizada y el dolor de cabeza son otros síntomas frecuentes. La debilidad,
la pérdida de peso, la palidez, las náuseas y el dolor abdominal también se han
relacionado con el feocromocitoma. Ocasionalmente, los pacientes con
incidentaloma suprarrenal o aquellos que se someten a exámenes periódicos de
detección de un síndrome familiar pueden estar asintomáticos. El cuadro 50.3
resume las indicaciones para la detección de feocromocitoma.

Recuadro 50.3. Detección de feocromocitoma.
Diagnóstico
Debido a que las catecolaminas (dopamina,
norepinefrina y epinefrina) por lo general se secretan de manera episódica y
tienen vidas medias cortas, los niveles aleatorios pueden pasar por alto el
diagnóstico a menos que se verifiquen durante un ataque paroxístico. Por lo
tanto, no se recomienda la medición de los niveles sanguíneos de catecolaminas
y puede ser engañoso. Debido a que el metabolismo de las catecolaminas
producidas por el feocromocitoma es en gran parte intratumoral, hay una
liberación continua de metabolitos Ometilados, es decir, metanefrina
(metabolitos de la epinefrina) y normetanefrina (metabolitos de la
norepinefrina). Como resultado, la medición de metanefrinas (que se refiere a
la combinación de metanefrina y normetanefrina) en sangre u orina se ha
convertido en la prueba de diagnóstico preferida.
La Endocrine Society recomienda el uso de metanefrinas
libres de plasma o metanefrinas libres de orina de 24 horas para la detección
inicial de feocromocitomas y paragangliomas. Las metanefrinas libres de plasma
tienen la ventaja de un simple análisis de sangre venoso periférico y aleatorio
en el consultorio. Las metanefrinas plasmáticas tienen un alto valor predictivo
negativo, excepto en pacientes con enfermedad preclínica temprana o tumores
secretores de dopamina. Por otro lado, una recolección de orina de 24 horas
para catecolaminas fraccionadas y metanefrinas también es una prueba confiable
para diagnosticar feocromocitoma. Los niveles de metanefrina en sangre u orina
que están 2 o 3 veces elevados por encima del límite superior normal se
consideran diagnósticos de feocromocitoma. Los medicamentos que interfieren,
los antidepresivos tricíclicos, la levodopa y otros simpaticomiméticos pueden
provocar elevaciones más pequeñas de falsos positivos en las metanefrinas. Si
la prueba es equívoca y la sospecha clínica de feocromocitoma es alta, se puede
considerar la prueba de supresión con clonidina.
Después de la confirmación bioquímica del exceso de
catecolaminas, la tomografía computarizada o la resonancia magnética del
abdomen generalmente diagnostican feocromocitoma suprarrenal porque la mayoría
de los tumores tienen un tamaño ≥2 a 3 cm. Se realizan imágenes funcionales,
como la gammagrafía con 123I-metayodobencilguanidina (MIBG), si las imágenes
suprarrenales son negativas para buscar un paraganglioma extraadrenal. MIBG se
absorbe en gránulos neurosecretores adrenérgicos y, por lo tanto, forma
imágenes de tumores de cromaina. Otras técnicas de imagen para localizar
tumores productores de catecolaminas incluyen gammagrafía con octreótido y
tomografía por emisión de positrones (PET) con 18F-luorodesoxiglucosa.
Tratamiento
La extirpación quirúrgica del feocromocitoma es el
tratamiento de elección, pero es fundamental tratar preoperatoriamente al
paciente con bloqueo alfa-adrenérgico. Los bloqueadores alfa-adrenérgicos
incluyen fenoxibenzamina, un fármaco no selectivo y no competitivo, o
bloqueadores alfa selectivos como la doxazosina o la prazosina. Los
bloqueadores beta no deben iniciarse antes de un bloqueo alfa adecuado porque
la estimulación de los receptores alfa sin oposición puede aumentar aún más la
presión arterial. Una vez que se ha establecido el bloqueo alfa, se pueden usar
bloqueadores beta para controlar la taquicardia o las arritmias. La metirosina,
un inhibidor de la síntesis de catecolaminas, puede usarse antes de la
operación en algunas situaciones. Se debe prescribir de forma rutinaria una
mayor expansión de volumen preoperatoria con una ingesta alta de sodio porque
se sabe que los pacientes con feocromocitoma tienen un volumen plasmático
reducido. Después de la resección de un feocromocitoma, los niveles de
catecolaminas deben medirse periódicamente para descartar una enfermedad
maligna.
Feocromocitoma y síndromes genéticos
El feocromocitoma y el paraganglioma ahora se han
atribuido a más de 15 mutaciones de la línea germinal. Debido a que se
encontrará que aproximadamente el 40% de todos estos tumores albergan una de
estas mutaciones, ahora se recomienda enfáticamente que todos los pacientes con
feocromocitoma o paraganglioma sean considerados para pruebas genéticas. Los
genes y síndromes asociados con el feocromocitoma y el paraganglioma incluyen
el síndrome de von HippelLindau (VHL), la neoplasia endocrina múltiple 2A y 2B,
la neuroibromatosis tipo 1, los genes de la succinato deshidrogenasa (A, B, C,
D y AF2), el gen TMEM127, el gen MAX , gen del factor 2-alfa inducible por
hipoxia y deficiencia de fumarato hidratasa. Muchos de estos síndromes
tumorales también aumentan el riesgo de desarrollar otros tumores y secuelas.
Los paragangliomas se localizan en la cabeza y el
cuello y también en el tórax, el abdomen y la pelvis. El cuadro 50.4 enumera
las mutaciones frecuentes de la línea germinal en el feocromocitoma y el
paraganglioma.

Box 50.4. Mutaciones de línea germinal en
feocromocitoma y paraganglioma.
Incidentaloma suprarrenal
Una masa suprarrenal >1 cm que se descubre por
casualidad en imágenes radiológicas se denomina incidentaloma suprarrenal. La
prevalencia de incidentalomas suprarrenales es mayor con la edad. La autopsia y
las series de imágenes han estimado que la prevalencia de masas suprarrenales
descubiertas incidentalmente es de entre 1% y 10%. Por el contrario, los
incidentalomas suprarrenales son poco comunes en pacientes menores de 30 años.
Cuando se identifica una masa suprarrenal, el enfoque requiere la evaluación
del potencial maligno (¿Es un tumor maligno o benigno o una entidad no
tumoral?) y el estado funcional (¿Esta masa hipersecreta hormonas suprarrenales?).
El primer paso en la evaluación debe ser una revisión
cuidadosa de la historia y el examen físico en busca de indicios de secreción
hormonal excesiva. Por lo general, se recomienda evaluar la hipersecreción de
cortisol en todos los tumores suprarrenales, por lo general a través de la prueba
de supresión con dexametasona (DST), de
1 mg, que es la más sensible. Se considera que los valores de cortisol
posteriores a la dexametasona >5 μg/dl representan un hipercortisolismo
manifiesto: si los signos o síntomas del síndrome de Cushing no son aparentes,
esto se conoce como hipercortisolismo subclínico o síndrome de Cushing
subclínico. También se considera que los valores de cortisol posdexametasona
entre 1,8 y 5 representan un hipercortisolismo subclínico o un
hipercortisolismo intermedio y están asociados con un mayor riesgo de
desarrollar resultados cardiovasculares y muerte. Tradicionalmente, los valores
de cortisol posdexametasona de <1.8 μg/dL se consideran "normales"
o representativos de un tumor suprarrenal "no funcional"; sin
embargo, la evidencia más reciente sugiere que incluso los tumores
suprarrenales “no funcionales” aumentan el riesgo de diabetes incidente. Aunque
en general se acepta que los tumores suprarrenales con síndrome de Cushing
manifiesto requieren una intervención urgente, hasta la fecha no existen
estudios de intervención o estudios prospectivos longitudinales con evidencia
que apoye las intervenciones para mitigar los resultados en el
hipercortisolismo subclínico. Además de la evaluación de hipercortisolismo, se
debe considerar la detección de feocromocitoma y AP, especialmente en pacientes
con hipertensión u otros signos clínicos que sugieran estos trastornos. Si se
observan características clínicas de hipersecreción de andrógenos en mujeres,
se deben medir los niveles de DHEAS.
La siguiente preocupación es si la masa es maligna.
Las características radiológicas que predicen la malignidad incluyen el tamaño
del tumor y el fenotipo de imagen. Las masas >6 cm tienen más probabilidades
de ser malignas y deben extirparse. Los adenomas suprarrenales benignos suelen
tener menos de 4 cm (generalmente 1 a 2 cm), son homogéneos con márgenes lisos
y característicamente ricos en lípidos según los criterios de CT o MRI. Los
adenomas suprarrenales benignos son ricos en lípidos, por lo general <10
unidades Hounsild en la tomografía computarizada sin contraste. Los adenomas
benignos también tienen un lavado rápido del medio de contraste en la
tomografía computarizada con contraste. Las lesiones de 4 a 6 cm se encuentran
en el área gris y la decisión de extirpar quirúrgicamente debe basarse en la
edad del paciente, el fenotipo de imagen y las condiciones coexistentes. Para
diagnosticar la enfermedad metastásica de una neoplasia maligna extraadrenal,
se puede utilizar una biopsia por aspiración con aguja en un paciente con
antecedentes de neoplasia maligna. debe realizarse solo después de descartar
feocromocitoma.
En resumen, para las lesiones que son hipersecretoras
o > 6 cm de diámetro, se debe realizar una resección quirúrgica. Para las
lesiones que probablemente sean benignas, se deben realizar estudios de imagen
repetidos a los 6, 12 y 24 meses. Un aumento de tamaño > 1 cm por año debe
generar preocupación por una posible malignidad y se debe considerar la
resección (Cuadro 50.5 y Fig. 50.4).

Box 50.5. Evaluación del incidentaloma suprarrenal
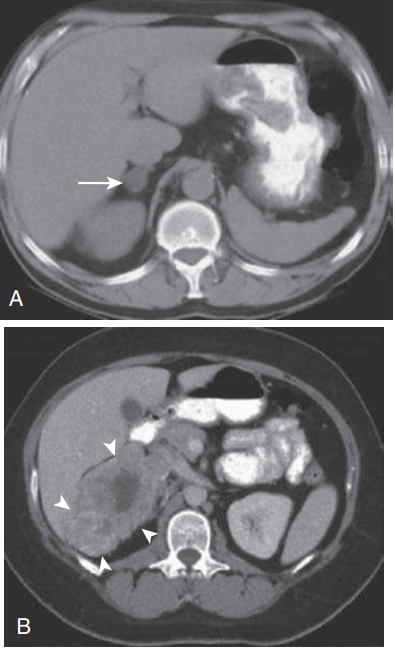
Figura 50.4. (A) Masa suprarrenal derecha de 1,5 cm
(flecha) que mide −8 unidades de campo Houns (HU) que se observa en esta
tomografía computarizada con contraste oral. (B) Tomografía computarizada con
contraste oral e intravenoso en un paciente con carcinoma suprarrenal. En
contraste con el tumor en (A), la masa suprarrenal derecha es grande (12 cm;
puntas de flecha) y heterogénea. La masa es contigua a la vena cava inferior y
se extiende hacia el lóbulo derecho del hígado y el polo superior del riñón.
(De Trikudanathan S, Dluhy RG. Disfunción cortical suprarrenal. En: Blake MA,
Boland G, eds. Adrenal Imaging. Nueva York: Humana Press; 2009:35–56.)
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (CAH) es un
trastorno autosómico recesivo que resulta de mutaciones de pérdida de función
de las enzimas involucradas en la síntesis de cortisol. La deficiencia
enzimática más frecuente es la deficiencia de 21-hidroxilasa. Los pacientes con
deficiencia de 21-hidroxilasa tienen una síntesis alterada de cortisol; como
resultado, la secreción compensatoria de ACTH da como resultado la derivación
de precursores hacia la vía de los andrógenos. El fenotipo varía desde
genitales ambiguos en niñas recién nacidas hasta hirsutismo en la edad adulta y
precocidad sexual en varones. La síntesis de aldosterona también se ve
alterada, lo que produce pérdida de sal, retraso en el crecimiento e
hipotensión.
En la CAH de inicio tardío, hay una deficiencia enzimática
parcial de 21-hidroxilasa que produce hirsutismo y oligomenorrea en mujeres
adultas. su diagnóstico se establece al documentar niveles matutinos elevados
de 17hidroxiprogesterona (17OHP) o un aumento anormal en los niveles de 17OHP
después de la estimulación con ACTH. El objetivo terapéutico en adultos con CAH
de inicio tardío es la dosis mínima de esteroides de acción prolongada para
suprimir la producción de andrógenos. Por lo general, se administran 5 mg de
prednisona o 0,25 a 0,5 mg de dexametasona al acostarse para suprimir la
producción de andrógenos adrenocorticales.
FUENTE:
The
Brigham Intensive Review of
Internal Medicine. (2022)
Ajay K. Singh, MBBS, FRCP, MBA
Joseph Loscalzo, MD, PhD
Bertagna X, Guignat L, Groussin L, et al. Cushing’s
disease. Best Pract
Res Clin Endocrinol Metab. 2009;23(5):607–623.
Chao CT, Wu VC, Kuo CC, et al. Diagnosis and
management of
primary aldosteronism: an updated review. Ann Med.
2013;45(4):
375–383.
Funder JW. Aldosterone, hypertension and heart
failure: insights
from clinical trials. Hypertens Res.
2010;33(9):872–875.
Gomez-Sanchez CE, Rossi GP, Fallo F, et al. Progress in primary
aldosteronism: present challenges and perspectives.
Horm Metab
Res. 2010;42(6):374–381.
Kannan S, Remer EM, Hamrahian AH. Evaluation of
patients with
adrenal incidentalomas. Curr Opin Endocrinol Diabetes
Obes.
2013;20(3):161–169.
Mulatero P, Monticone S, Bertello C, et al. Evaluation
of primary
aldosteronism. Curr Opin Endocrinol Diabetes Obes.
2010;17(3):
188–193.
Neary N, Nieman L. Adrenal insuiciency: etiology,
diagnosis and
treatment. Curr Opin Endocrinol Diabetes Obes.
2010;17(3):
217–223.
Nieman LK. Approach to the patient with an adrenal
incidentaloma.
J Clin Endocrinol Metab. 2010;95(9):4106–4113.
Pelosof LC, Gerber DE. Paraneoplastic syndromes: an
approach to
diagnosis and treatment. Mayo Clin Proc.
2010;85(9):838–854.
Willatt JM, Francis IR. Radiologic evaluation of
incidentally discov ered adrenal masses. Am Fam Physician.
2010;81(11):1361–1366.