El pioderma gangrenoso (PG) es una dermatosis
neutrofílica poco común que se presenta como un trastorno inflamatorio y
ulcerativo de la piel. A diferencia de su nombre, el PG no es una afección
infecciosa ni gangrenosa. La presentación más común del PG es una pápula o
pústula inflamatoria que progresa a una úlcera dolorosa con un borde socavado
violáceo y una base purulenta ( imagen 1A-G ). El PG también puede presentar
lesiones ampollosas, vegetativas, periestomales y extracutáneas.

Imagen 1A. Pioderma gangrenoso.
Se presenta una úlcera purulenta en la extremidad.

Imagen 1B. Pioderma gangrenoso.
Múltiples úlceras de borde violáceo en miembro inferior.

Imagen 1C. Pioderma gangrenoso.
Úlceras de borde violáceo en pie y tobillo.

Imagen 1 D. Pioderma gangrenoso.
Esta úlcera purulenta con un borde irregular y
violáceo es compatible con pioderma gangrenoso.

Imagen 1 E. Pioderma gangrenoso.
En la extremidad inferior se presenta una úlcera
grande con bordes socavados y base purulenta.

Imagen 1 F. Pioderma gangrenoso .
En la extremidad inferior se presenta una úlcera
grande con bordes socavados y base purulenta.

Imagen 1G. Pioderma gangrenoso.
Múltiples lesiones activas y curativas de pioderma
gangrenoso con cicatrización cribiforme en paciente con enfermedad inflamatoria
intestinal.
Más de la mitad de los pacientes con PG desarrollan el trastorno asociado a una enfermedad sistémica subyacente. La enfermedad inflamatoria intestinal, los trastornos hematológicos y la artritis representan las comorbilidades más frecuentes.
El diagnóstico de PG se basa en el reconocimiento de
hallazgos clínicos e histológicos consistentes y la exclusión de otros
trastornos cutáneos inflamatorios o ulcerativos. No existen hallazgos
patognomónicos, clínicos ni histológicos de PG.
EPIDEMIOLOGÍA
La PG es un trastorno poco frecuente con una
incidencia estimada de 3 a 10 casos por millón de personas al año [ 1 ]. Pueden
verse afectadas personas de cualquier edad, incluidos niños [ 2-4 ]. La PG se
presenta con mayor frecuencia en adultos jóvenes y de mediana edad, con una
edad promedio de inicio entre los 40 y los 60 años [ 5-8 ]. En cuanto a sexos,
las mujeres se ven afectadas con mayor frecuencia [ 7 ].
PATOGENIA
En consonancia con su clasificación como dermatosis
neutrofílica, la PG se caracteriza por infiltrados con predominio de
neutrófilos en la piel. Se han comenzado a investigar las causas de la
desregulación de la respuesta inmunitaria innata y adaptativa, además de los
factores genéticos, en la patogénesis de la PG.
los estudios han revelado
perfiles complejos de expresión génica en la piel de pacientes con PG. La piel
lesionada de PG ha aumentado la expresión de interleucina (IL) 1-beta,
IL-1-alfa, IL-8, IL-12, IL-15, IL-17, IL-23, IL-36 y factor de necrosis tumoral
(TNF)-alfa [ 9-11 ]. Esta regulación positiva conduce a un estado
proinflamatorio a través de una mayor activación de inflamasomas, desregulación
del sistema inmunitario innato y reclutamiento y activación de neutrófilos [ 9
]. El tejido de PG también ha aumentado la expresión de receptores de
reconocimiento de patrones (PRR), JAK2 y STAT1 , lo que implica una disfunción
de la respuesta inmunitaria adaptativa e innata [ 10 ]. La vía del complemento,
en particular C5a y su papel en la activación de neutrófilos, la quimiotaxis y
la señalización inflamatoria, también puede contribuir a la compleja
patogénesis de PG [ 12 ].
Factores genéticos: La susceptibilidad genética
probablemente también contribuya al desarrollo de PG. Se han reportado casos
familiares de PG [ 13-16 ]. Además, el síndrome PAPA (artritis piógena,
pioderma gangrenoso y acné) (OMIM #604416), un trastorno autosómico dominante,
se ha vinculado a mutaciones en el gen PSTPIP1 / CD2BP1. Esta mutación provoca
una disminución de la inhibición del inflamasoma y, en última instancia, un
aumento de la producción de IL-1-beta y autoinflamación [ 17 ]. Se han
demostrado mutaciones de los genes autoinflamatorios MEFV , NLRP3 , NLRP12 ,
NOD2 y LPIN2 en PG, síndrome PASH (pioderma gangrenoso, acné e hidradenitis
supurativa) y otras afecciones inflamatorias, incluidas la fiebre mediterránea
familiar, el síndrome periódico asociado a la criopirina, la enfermedad de
Crohn y el síndrome SAPHO (sinovitis, acné, pustulosis, hiperostosis, osteítis)
[ 9 ].
Se han reportado errores congénitos adicionales en la inmunidad innata en PG, incluyendo mutaciones genéticas en BTK , IL1RN , ITGB2 , NFkB1 , PMSB8 , PLCG2 , RAG1 , TTC37 y WDR1 ; deficiencias del componente 2 del complemento/componente 4 del complemento (C2/C4) y del componente 7 del complemento (C7); y deficiencia de adhesión leucocitaria tipo 1 [ 18,19 ]. En una revisión sistemática, las mutaciones en PSMB8 , NLRP3 e IL1RN se asociaron con un curso más grave y atípico de PG; las mutaciones en RAG1 tuvieron las características clínicas más leves; y NFkB1 , ITGB2 y PSTPIP1 tuvieron las presentaciones más heterogéneas de PG [ 18 ].
Un mayor esclarecimiento de las vías moleculares en el
PG ha permitido el desarrollo de tratamientos más específicos, como los
inhibidores del TNF-alfa, IL-1, IL-17, IL-23 e IL-6.
MANIFESTACIONES CLÍNICAS
Las manifestaciones clínicas de la PG son variables y
pueden dividirse en cuatro subtipos principales: PG ulcerosa (clásica) (la
variante más común), PG ampollosa (atípica), PG pustulosa y PG vegetativa [ 20
].
Características comunes: Los subtipos de PG comparten una evolución
clínica caracterizada por la aparición de una pápula, pústula, vesícula o
nódulo inflamatorio que posteriormente se expande y se descompone para formar
una erosión o úlcera. Con la excepción del PG vegetativo, la lesión suele desarrollarse
rápidamente y el dolor suele ser mayor de lo esperado según la apariencia de la
úlcera [ 20 ]. Puede presentarse fiebre [ 21 ].
Patergia, un término usado para describir la inducción
o exacerbación de PG en sitios de trauma incidental o iatrogénico, se menciona
con frecuencia en las descripciones del comportamiento clínico de PG. Sin
embargo, no todos los pacientes con PG presentan esta característica. Los datos
sobre la proporción de pacientes con PG que experimentan patergia son
limitados. En un estudio retrospectivo de 103 pacientes con PG, se documentó
patergia en los registros médicos del 31 por ciento de los pacientes [ 7 ]. En
una serie de 166 pacientes con PG, 25 (15 por ciento) experimentaron 33
episodios de recurrencia posquirúrgica o exacerbación de la enfermedad; los
factores asociados con un mayor riesgo de patergia incluyeron procedimientos
quirúrgicos abiertos, cirugía de Mohs y PG presente en el momento del
procedimiento [ 22 ].
Subtipos: A
continuación se proporcionan descripciones de las principales variantes de PG:
Pioderma gangrenoso ulcerativo (clásico): La variante
ulcerativa del PG representa la gran mayoría de los casos. El PG ulcerativo
suele comenzar como una pápula, pústula o vesícula dolorosa e inflamatoria que
se desarrolla en piel de aspecto normal o en un lugar de traumatismo ( imagen
2A-B ). Las extremidades inferiores y el tronco son los sitios de afectación
más comunes, aunque pueden presentarse lesiones en otras zonas [ 1 ].
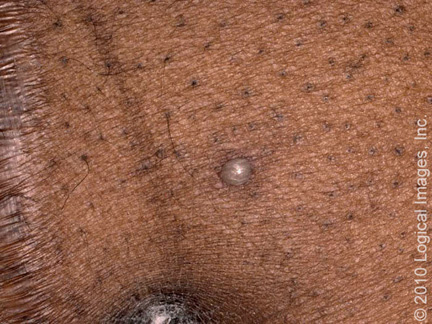
Imagen 2A. Pioderma gangrenoso
Esta pústula sobre base eritematosa representa una
lesión temprana de pioderma gangrenoso.

Imagen 2B. Pioderma gangrenoso.
Lesión temprana en pioderma gangrenoso que se presenta
como una placa pustulosa y violácea con descomposición incipiente.
La lesión inflamatoria inicial se expande
posteriormente periféricamente y degenera centralmente, dando lugar a la
formación de una úlcera ( imagen 1A , imagen 1B y imagen 1C-G ). El borde de la
úlcera suele presentar una coloración azulada o violácea y un aspecto socavado.
La expansión irregular del tejido afectado puede dar lugar a una configuración
serpiginosa. La base de la úlcera es purulenta y necrótica, y su profundidad
suele extenderse hasta la grasa subcutánea y, en ocasiones, alcanza la fascia [
1 ].
Los pacientes pueden presentar una sola lesión o
múltiples lesiones en diversas etapas de desarrollo. Las úlceras suelen
resolverse con cicatrices atróficas y cribiformes.
Pioderma gangrenoso ampolloso (atípico): el pioderma
gangrenoso ampolloso es una variante superficial menos común del PG que se
observa con mayor frecuencia en pacientes con PG relacionado con una enfermedad
hematológica [ 6 ]. A diferencia del pioderma ulcerativo, los brazos y la cara
son los sitios de afectación más comunes [ 1,6 ]. Los pacientes suelen
presentar el rápido desarrollo de ampollas inflamatorias de color azul grisáceo
en las áreas afectadas [ 6 ]. Las ampollas se erosionan rápidamente, dando
lugar a úlceras superficiales ( imagen 3 ).

Imagen 3. Pioderma gangrenoso bulloso o ampolloso.
Tres grandes ampollas hemorrágicas en el dorso de la
mano y los dedos. La ampolla del dedo índice se ha roto, lo que ha provocado
una gran erosión con un borde pustuloso irregular y elevado, y un exudado
hemorrágico purulento.
Debido a la fuerte asociación entre la PG ampollosa (bullosa),
y la enfermedad hematológica, los pacientes que no presentan un trastorno
hematológico asociado deben ser seguidos de cerca para detectar el desarrollo
de un trastorno hematológico [ 23 ].
Pioderma gangrenoso pustuloso: el PG pustuloso suele
presentarse en pacientes con enfermedad inflamatoria intestinal y tiende a
surgir durante períodos de exacerbaciones agudas de la enfermedad intestinal [
20 ]. Los pacientes afectados presentan un rápido desarrollo de pústulas
dolorosas rodeadas de eritema ( imagen 4 ). La fiebre y las artralgias
concomitantes son comunes [ 1 ]. La pioestomatitis vegetante, que se presenta
con múltiples pústulas pequeñas y erosiones en la mucosa oral, puede ser una
variante del PG pustuloso [ 1,24 ].

Imagen 4. Pioderma gangrenoso pustuloso.
Se observan múltiples pústulas con eritema periférico
compatible con pioderma gangrenoso pustuloso en la extremidad. También se
observan máculas hemorrágicas dispersas.
Pioderma gangrenoso vegetativo: El PG vegetativo
(también conocido como pioderma granulomatoso superficial) es una forma de PG
típicamente localizada, solitaria y superficial que se presenta como un nódulo,
placa o úlcera indolente y ligeramente dolorosa ( imagen 5 ). Suele presentar
un aspecto verrugoso. Los bordes socavados y las bases purulentas del PG
ulcerativo están ausentes. La cabeza, el cuello y el tronco son los sitios más
comunes de PG vegetativo [ 1 ].

Imagen 5. Pioderma gangrenoso vegetante.
En el tobillo se presenta una úlcera con superficie
verrugosa.
Sitios especiales: Además de los subtipos
morfológicos mencionados anteriormente, se han utilizado otros términos para
describir ciertas presentaciones de PG. Algunos ejemplos son PG periostomal, PG
genital y PG extracutáneo.
Pioderma gangrenoso periostomal: El pioderma gangrenoso
periostomal es una manifestación poco común de pioderma gangrenoso que se
caracteriza por el desarrollo de lesiones compatibles con pioderma gangrenoso
ulcerativo adyacentes a un estoma ( imagen 6 ). Esto suele ocurrir en pacientes
con colitis ulcerosa o enfermedad de Crohn y puede desarrollarse meses o
incluso décadas después de la colocación de un estoma. El traumatismo causado
por el mantenimiento del estoma o la irritación por las secreciones estomatosas
pueden influir en el desarrollo de pioderma gangrenoso periostomal [ 1 ].

Imagen 6. Pioderma gangrenoso periostomal.
El pioderma gangrenoso periestomal es causado por un
proceso inflamatorio que produce ulceraciones cutáneas graves y dolorosas.
Pioderma gangrenoso genital: el pioderma gangrenoso
genital puede afectar la vulva, el pene o el escroto. Las lesiones se asemejan
a las del pioderma gangrenoso ulcerativo.
Pioderma gangrenoso extracutáneo: En raras ocasiones,
el PG afecta localizaciones extracutáneas. En estos casos, se desarrolla una
infiltración neutrofílica estéril en zonas como pulmones, intestino, córnea,
hígado, bazo, corazón, huesos, músculos y sistema nervioso central [ 25-37 ].
Pioderma gangrenoso posoperatorio: El PG
posoperatorio es la aparición de PG en la zona quirúrgica, generalmente dentro
de las dos semanas posteriores a la cirugía [ 38 ]. Los síntomas iniciales son
eritema en la zona quirúrgica y dolor intenso, desproporcionado a la
exploración física, seguido de dehiscencia de la herida o la aparición de
ulceraciones puntiformes que se fusionan para formar úlceras más grandes. El PG
posoperatorio parece ser más común en mujeres que en hombres. Las zonas de
afectación más comunes son las mamas y el abdomen [ 38 ].
TRASTORNOS ASOCIADOS
Más del 50 % de los pacientes con PG presentan una
enfermedad sistémica asociada, con mayor frecuencia enfermedad inflamatoria
intestinal, artritis y enfermedad hematológica o neoplasia hematológica maligna
[ 5-7 ]. La proporción de pacientes con comorbilidades específicas varía entre
los estudios [ 5-7,39 ]. En uno de los estudios más amplios, un estudio de
cohorte retrospectivo de 356 adultos con PG, 161 (45 %) presentaron
comorbilidades asociadas, incluyendo [ 39 ]:
- 146 pacientes (41 por ciento) con enfermedad inflamatoria intestinal
- 73 pacientes (21 por ciento) con artritis inflamatoria
- 23 pacientes (7 por ciento) con neoplasias malignas de órganos sólidos
- 21 pacientes (6 por ciento) con neoplasias malignas hematológicas
- 17 pacientes (5 por ciento) con trastornos hematológicos (gammapatía monoclonal de significado incierto, síndrome mielodisplásico o policitemia vera)
Hallazgos adicionales de este estudio sugieren que la
edad del paciente influye en el riesgo de comorbilidades específicas. Los
pacientes de 65 años o más presentaron mayor probabilidad de presentar artritis
reumatoide, espondilitis anquilosante, neoplasias malignas de órganos sólidos,
neoplasias malignas hematológicas y trastornos hematológicos asociados que los
pacientes más jóvenes. La enfermedad inflamatoria intestinal fue más frecuente
en pacientes menores de 65 años.
La PG puede preceder o seguir al diagnóstico de un
trastorno asociado y puede o no ser paralela al curso clínico de la enfermedad
asociada [ 23,40 ].
La PG puede presentarse como una característica de
síndromes autoinflamatorios, como el síndrome PAPA (artritis piógena, pioderma
gangrenoso y acné), el síndrome PAPASH (artritis piógena, pioderma gangrenoso,
acné e hidradenitis supurativa [hidradenitis supurativa]) y el síndrome PASH
(pioderma gangrenoso, acné e hidradenitis supurativa) [ 41-44 ]. El síndrome
PAPA es un trastorno autosómico dominante y se presenta de manera secundaria a
un defecto en el gen PSTPIP1 / CD2BP1 . El síndrome PAPASH se ha descrito en un
adolescente en quien se detectó una nueva mutación en el gen PSTPIP1 [ 41 ].
También se ha descrito la PG en un paciente con celulitis disecante del cuero cabelludo,
hidradenitis supurativa y acné inflamatorio [ 45 ].
Los hallazgos similares a PG también pueden
presentarse en el síndrome autoinflamatorio VEXAS (vacuolas, enzima E1, ligado
al cromosoma X, autoinflamatorio y somático), y el síndrome VEXAS que presenta
lesiones similares a PG puede no haberse reconocido antes de la aparición de
este trastorno. En una revisión sistemática de estudios que incluyó a pacientes
con diagnóstico de PG asociado a síndrome mielodisplásico o leucemia
mielomonocítica crónica que no se sometieron a pruebas genéticas confirmatorias
para el síndrome VEXAS, 29 de 53 pacientes presentaron características
sugestivas del síndrome VEXAS [ 46 ]. Todos los pacientes eran varones, la
mayoría presentaba síntomas sistémicos asociados y la enfermedad refractaria
fue frecuente.
Entre los ejemplos de trastornos adicionales que se
han vinculado con poca frecuencia a la PG se incluyen la enfermedad pulmonar,
el lupus eritematoso sistémico, la enfermedad tiroidea, los cánceres de órganos
sólidos, la hepatitis viral y autoinmune, la sarcoidosis, la depresión mayor y
la diabetes [ 7,40,47 ]. Además, también se ha informado de PG en pacientes
expuestos a levamisol y rituximab [ 48,49 ]. Se necesitan más estudios para
validar la relación entre estos trastornos y la PG.
DIAGNÓSTICO
Los hallazgos clínicos, histopatológicos y de
laboratorio en el PG son inespecíficos, y el diagnóstico de PG solo puede
realizarse una vez descartadas otras posibilidades diagnósticas. Se debe
realizar al menos una historia clínica, una exploración física y una biopsia
cutánea a todos los pacientes [ 40 ]. En pacientes con enfermedades asociadas
al PG, la aparición de una úlcera debe hacer sospechar este diagnóstico.
Además, una vez diagnosticado el PG, se debe evaluar a los pacientes para
detectar la presencia de una enfermedad subyacente asociada.
Criterios diagnósticos del pioderma gangrenoso
ulcerativo clásico
Criterios diagnósticos: Históricamente, no se han contado con
criterios diagnósticos universalmente aceptados y validados para la PG. Los
criterios diagnósticos publicados en 2004 exigen la exclusión de todos los
demás diagnósticos, lo que puede resultar complejo o poco práctico en el ámbito
clínico ( tabla 1 ) [ 50 ].

Tabla 1. Criterios diagnósticos propuestos para el
pioderma gangrenoso ulcerativo clásico (2004)
Los criterios diagnósticos más recientes para la PG
ulcerosa, desarrollados mediante un consenso Delphi de expertos
internacionales, proporcionan una mayor estructura al proceso diagnóstico y son
nuestra herramienta preferida para el diagnóstico [ 51 ]. Estos criterios
incluyen un único criterio principal y ocho criterios secundarios, que incluyen
hallazgos histológicos, de la anamnesis, de la exploración clínica y del
tratamiento.
Criterio principal:
- Biopsia del borde de la úlcera que muestra un infiltrado neutrofílico
Criterios menores:
- Exclusión de infección
- Patergia
- Antecedentes personales de enfermedad inflamatoria intestinal o artritis inflamatoria
- Antecedentes de pápula, pústula o vesícula que se ulceró rápidamente
- Eritema periférico, borde socavado y dolor en el sitio de la ulceración.
- Ulceraciones múltiples (al menos una de ellas en la parte anterior de la parte inferior de la pierna)
- Cicatrices cribiformes o de "papel arrugado" en sitios de úlceras curadas
- Disminución del tamaño de la úlcera al mes de iniciar la medicación inmunosupresora.
Para el diagnóstico son necesarios al menos el
criterio mayor y cuatro criterios menores.
Un tercer criterio diagnóstico, la puntuación
PARACELSUS, utiliza un sistema de puntos con criterios mayores y menores
ligeramente diferentes [ 52 ]. Este sistema fue diseñado específicamente con el
propósito de diferenciar las úlceras venosas de la pierna de la PG.
Los criterios de diagnóstico se utilizan
predominantemente para estudios clínicos, pero pueden ofrecer un marco para el
diagnóstico en la práctica diaria.
Evaluación clínica:
La evaluación clínica del paciente con una lesión sospechosa de PG debe
incluir una historia clínica y una exploración física exhaustivas. Se debe
interrogar al paciente sobre el desarrollo de la lesión, los síntomas asociados
y sus antecedentes médicos personales. Algunos ejemplos de hallazgos que se
pueden obtener a través de la historia clínica del paciente y que respaldan el
diagnóstico de PG son:
- Curso rápido de desarrollo de la lesión
- Lesión inicial consistente con pápula, pústula o vesícula.
- Dolor desproporcionado a la lesión
- Historia de trauma previo (patergia)
- Historial de trastornos asociados con PG (ver 'Trastornos asociados' más arriba)
El examen físico es útil para identificar la extensión
de la enfermedad, limitar el diagnóstico diferencial y detectar signos clínicos
de trastornos asociados a PG.
Biopsia: Dado
que los hallazgos histopatológicos del PG son inespecíficos, las biopsias son
especialmente útiles para descartar otros trastornos que puedan presentar
hallazgos clínicos similares. La biopsia está indicada en pacientes sin
antecedentes de PG y en pacientes con PG establecido que presentan lesiones con
características atípicas o que no responden al tratamiento según lo previsto.
Si bien los médicos suelen preocuparse por la patergia inducida por la biopsia,
la necesidad de descartar otros trastornos y asegurar un tratamiento adecuado
prevalece sobre la preocupación por la inducción de una exacerbación del PG.
Procedimiento:
El procedimiento óptimo para la biopsia es una biopsia incisional
elíptica que abarca tanto el borde de la lesión inflamada como el borde de la
úlcera y se extiende verticalmente hacia la grasa subcutánea [ 50 ]. Se debe
enviar una muestra de tejido que incluya el borde inflamado para un examen
histopatológico de rutina y tinciones microbianas. Además, se debe enviar una
muestra de la úlcera para cultivo a fin de evaluar infecciones bacterianas,
fúngicas y micobacterias atípicas.
Patología: Las
muestras tomadas de las lesiones iniciales de PG muestran inflamación
perifolicular y formación de abscesos intradérmicos ( imagen 8A-B ) [ 53 ]. Una
vez que las lesiones progresan a ulceración, se suele detectar necrosis
epidérmica y dérmica superficial con un infiltrado de células inflamatorias
mixtas subyacente y formación de abscesos. Se pueden encontrar células gigantes,
y el tejido del borde de la lesión puede mostrar cambios vasculares sugestivos
de vasculitis linfocítica. También puede presentarse vasculitis
leucocitoclástica [ 53 ].

Imagen 8A Pioderma gangrenoso.
En la dermis se observa un denso infiltrado
inflamatorio con predominio de neutrófilos.

Imagen 8 B. Pioderma gangrenoso
Infiltrado neutrofílico denso en el pioderma
gangrenoso.
Se pueden identificar hallazgos adicionales según el
subtipo de PG. Las muestras de PG ampolloso presentan ampollas subepidérmicas.
La hiperplasia pseudoepiteliomatosa, los trayectos fistulosos y los granulomas
en empalizada son características compatibles con PG vegetativo.
Los estudios de inmunofluorescencia directa presentan
hallazgos inespecíficos en la PG. Se puede detectar el depósito de IgM, C3 y
fibrina en las paredes vasculares [ 40 ]. Debido a la naturaleza inespecífica
de los resultados, la inmunofluorescencia directa suele realizarse solo cuando
hallazgos clínicos adicionales indican la necesidad de descartar enfermedad
ampollosa autoinmune, lupus o vasculitis como causa de ulceración [ 50 ].
Estudios de laboratorio: Al igual que las biopsias de tejido, no
existen estudios de laboratorio que proporcionen un diagnóstico definitivo de
PG. Pueden presentarse hallazgos inespecíficos como leucocitosis, aumento de la
velocidad de sedimentación globular (VSE) y aumento de la proteína C reactiva [
21 ].
Las pruebas de laboratorio son muy útiles para reducir
el diagnóstico diferencial e identificar la presencia de enfermedades asociadas
a la PG. Las recomendaciones para la evaluación diagnóstica de estos pacientes
varían. Generalmente, realizamos los siguientes estudios en pacientes con
hallazgos clínicos e histopatológicos compatibles con PG:
- Hemograma completo : para evaluar trastornos hematológicos subyacentes.
- Panel metabólico completo : para evaluar disfunción hepática o renal y anomalías de la glucosa antes de iniciar el tratamiento con glucocorticoides sistémicos o agentes inmunosupresores.
- Título de anticuerpos antinucleares : para evaluar la presencia de lupus eritematoso sistémico o trastornos vasculares del colágeno asociados con PG.
- Anticuerpos citoplasmáticos antineutrófilos : para evaluar la vasculitis granulomatosa como causa de ulceración.
- Estudios de hipercoagulabilidad : detección de anticuerpos antifosfolípidos para evaluar el síndrome antifosfolípido como causa de ulceración; según la sospecha clínica, otras pruebas para evaluar estados trombóticos (por ejemplo, crioglobulinas, factor V Leiden, metilen tetrahidrofolato reductasa).
- Panel de hepatitis : para evaluar la presencia de hepatitis B o C asociada, en particular en pacientes en quienes se considera una terapia inmunomoduladora.
- Factor reumatoide – Como componente de la evaluación de la crioglobulinemia y la artritis reumatoide.
- Electroforesis de inmunofijación sérica : para evaluar paraproteínas.
- Radiografía de tórax : para evaluar la presencia de compromiso extracutáneo y posible infección antes de iniciar la terapia inmunosupresora.
- Colonoscopia: para evaluar si existe enfermedad inflamatoria intestinal subyacente, a menos que se identifique otra causa de PG.
Se solicitan pruebas adicionales para evaluar
trastornos asociados y trastornos en el diagnóstico diferencial según la
sospecha clínica de enfermedades específicas. Las pruebas genéticas siguen
evolucionando y podrían orientar futuras terapias dirigidas.
Diagnóstico diferencial: Debido a la falta de una prueba definitiva
para el PG, puede haber diagnósticos erróneos. En un estudio retrospectivo, 15
de 157 pacientes consecutivos atendidos en una institución académica con
diagnóstico de PG (10 %) presentaron posteriormente un diagnóstico alternativo
[ 54 ].
Trastornos de oclusión vascular, enfermedad venosa,
vasculitis, malignidad, infección cutánea, fármacos, lesión tisular exógena y
trastornos inflamatorios ulcerativos (p. ej., enfermedad de Crohn cutánea y
necrobiosis lipoídica ulcerativa) pueden confundirse con PG [ 54 ]. El síndrome
de anticuerpos antifosfolípidos es uno de los trastornos más comunes que se
confunden con PG; otros trastornos que se confunden más comúnmente incluyen
úlceras por estasis venosa, granulomatosis con poliangeítis, úlceras facticias,
esporotricosis, poliarteritis nodosa, vasculopatía livedoide, linfoma de
células T angiocéntrico y crioglobulinemia [ 54 ]. La fascitis necrotizante es
un diagnóstico erróneo potencial adicional [ 55 ]. El desbridamiento agresivo
para este presunto diagnóstico puede conducir a una morbilidad y deformidad
significativas [ 56-58 ].
La historia clínica, la biopsia y los estudios de
laboratorio dirigidos obtenidos durante la evaluación del paciente ayudan a
distinguir el PG de otras enfermedades. Se recomienda reevaluar el diagnóstico
de PG si la enfermedad no responde al tratamiento según lo previsto.
TRATAMIENTO Y PRONÓSTICO
PRINCIPIOS DE TRATAMIENTO
En general, tras el diagnóstico de PG, los pacientes
reciben tratamiento con una combinación de terapias farmacológicas que suprimen
el proceso inflamatorio y medidas de cuidado de la herida que optimizan el
entorno para la cicatrización. Si bien los signos iniciales de mejoría pueden
ser evidentes a los pocos días de iniciar algunos tratamientos, a menudo se
requieren semanas o meses para lograr la cicatrización completa de la úlcera [
1,2 ].
Los principios principales del tratamiento incluyen:
- Datos limitados sobre eficacia: Debido a la escasez de datos de alta calidad sobre intervenciones para la PG, no existen directrices definitivas para el manejo de los pacientes. El enfoque terapéutico de la PG se basa principalmente en estudios pequeños no controlados e informes de experiencia clínica [ 3 ].
- Influencia de la gravedad de la enfermedad en la selección del tratamiento inicial: La gravedad de la PG influye en la elección del tratamiento inicial. Si bien las terapias tópicas inducen la curación con éxito en algunos pacientes con enfermedad limitada, los pacientes con PG más extensa generalmente requieren terapia sistémica para detener la actividad de la enfermedad.
- Función de la terapia sistémica adyuvante: La administración sistémica de glucocorticoides o ciclosporina es nuestro tratamiento inicial preferido para la PG extensa. Sin embargo, la monoterapia puede ser insuficiente, y el tratamiento a largo plazo con estos fármacos se asocia con el riesgo de efectos adversos graves. Con frecuencia incorporamos otros fármacos inmunomoduladores sistémicos (p. ej., infliximab , adalimumab , micofenolato de mofetilo) para reducir la exposición a los glucocorticoides sistémicos y la ciclosporina y promover la curación de la PG.
- Impacto de las enfermedades concomitantes asociadas al PG: El PG puede presentarse en asociación con diversos trastornos, como la enfermedad inflamatoria intestinal, la artritis inflamatoria, las neoplasias malignas y las enfermedades hematológicas. La presencia de un trastorno asociado suele influir en la selección de tratamientos complementarios o posteriores para el PG, lo que contribuye a la selección de tratamientos beneficiosos tanto para el PG como para la enfermedad asociada.
- Evaluación de la respuesta al tratamiento: La estabilización de la progresión de la enfermedad y la reducción del dolor y de los signos visibles de inflamación (p. ej., coloración violácea en el borde de la herida) son signos tempranos importantes de respuesta. Posteriormente, la base de la herida comienza a llenarse de tejido de granulación y el diámetro de la herida disminuye progresivamente. La documentación del tamaño, número y ubicación de las úlceras, así como la toma de fotografías clínicas seriadas, son útiles para el seguimiento de la respuesta al tratamiento.
- Manejo de heridas: El cuidado de las heridas es un componente importante del manejo de la PG. Los principios clave incluyen el vendaje de las heridas, la monitorización de infecciones, la prevención de traumatismos y el uso prudente de la cirugía. (Véase "Manejo de heridas" a continuación).
SELECCIÓN DEL TRATAMIENTO
Terapia inicial:
La terapia con corticosteroides tópicos es nuestro tratamiento inicial
preferido para la PG limitada. El cuidado de la herida y el manejo del dolor
son componentes importantes del tratamiento.
Corticosteroides tópicos
La inyección intralesional de corticosteroides (p.
ej., acetónido de triamcinolona ) también se ha utilizado para el PG. Sin
embargo, algunos médicos, entre los que nos incluimos, evitan las inyecciones
intralesionales debido a la preocupación por la inducción de patergia. Se han
utilizado concentraciones de triamcinolona entre 6 y 40 mg/ml [ 4 ]. El
corticosteroide se inyecta circunferencialmente en la periferia de la úlcera.
La atrofia cutánea, la hipopigmentación y la supresión
del eje hipotálamo-hipofisario son ejemplos de efectos adversos de la terapia
con corticosteroides tópicos e intralesionales. Los efectos adversos se
analizan con más detalle por separado. (Véase "Corticosteroides tópicos:
Uso y efectos adversos", sección "Efectos adversos" y
"Inyección de corticosteroides intralesionales", sección
"Efectos adversos y riesgos" ).
Eficacia: Si bien los corticosteroides tópicos se
prescriben con frecuencia como terapia primaria y complementaria para la PG,
los datos sobre la eficacia de la terapia local con corticosteroides son
limitados [ 5-8 ]. Algunos ejemplos de estudios que evalúan la eficacia
incluyen:
Un estudio de cohorte prospectivo multicéntrico que
evaluó los resultados de la terapia tópica en 66 pacientes con PG apoya el
beneficio del tratamiento tópico con corticosteroides [ 7 ]. La mayoría de los
pacientes (74%) recibieron propionato de clobetasol al 0,05%; los pacientes
restantes recibieron ungüento tópico de tacrolimus al 0,03% o al 0,1%. A los
seis meses, 28 de los 64 pacientes (44%) para quienes se disponía de
información sobre la curación habían cicatrizado solo con terapia tópica,
incluidos 20 de 47 (43%) tratados con clobetasol y 5 de 10 (50%) tratados con
tacrolimus tópico. La mediana de tiempo hasta la curación fue de 145 días. El
tamaño inicial de la úlcera fue un predictor del tiempo hasta la curación.
En un estudio de cohorte prospectivo que incluyó a 49
pacientes con PG tratados con propionato de clobetasol al 0,05%, 20 (43 por
ciento) lograron la curación en un plazo de seis meses [ 7 ]. El tiempo medio
de curación fue de 136 días.
El beneficio de la terapia con corticosteroides
intralesionales en PG está documentado en informes de casos [ 6,9-12 ].
Respuesta insuficiente al tratamiento inicial: cuando la terapia con corticosteroides
tópicos no mejora la PG limitada en un plazo de dos a cuatro semanas, solemos
cambiar a ungüento de tacrolimus al 0,1 % o iniciar dapsona o minociclina oral
( algoritmo 1 ). Aunque los datos de eficacia son limitados, los perfiles de
efectos adversos relativamente favorables de estas terapias nos instan a
probarlas antes de proceder con terapias inmunosupresoras. (Véase
"Enfermedad más extensa y enfermedad limitada resistente al
tratamiento" más adelante).
Tacrolimus tópico
Administración y precauciones: El tacrolimus tópico ,
un inhibidor tópico de la calcineurina, se comercializa en pomada al 0,03 % y
al 0,1 %. Generalmente, recetamos pomada de tacrolimus al 0,1 % e indicamos a
los pacientes que la apliquen en la periferia de la úlcera inflamada una o dos
veces al día. Los signos de mejoría pueden ser evidentes en los primeros días o
semanas de tratamiento. Sin embargo, pueden ser necesarias varias semanas o
meses de tratamiento para la cicatrización completa de la úlcera [ 13-15 ].
El tacrolimus tópico suele tolerarse bien;
ocasionalmente, los pacientes experimentan una leve sensación de ardor en el
lugar de aplicación [ 14 ].
Eficacia – El tacrolimus tópico en concentraciones de
0,03% a 0,3% ha demostrado eficacia para PG en múltiples informes de casos,
algunas series de casos prospectivos y estudios no controlados [ 5,7,13,16 ].
En un estudio no controlado, una formulación compuesta de tacrolimus al 0,3% en
una pasta de carmelosa sódica pareció ser eficaz para PG periostomal [ 8 ].
Siete de 11 pacientes tratados con la formulación de tacrolimus y 5 de 13
pacientes tratados con loción o pomada de clobetasol al 0,05% lograron una
curación completa. En un estudio de cohorte prospectivo que incluyó a 10
pacientes con PG tratados con tacrolimus tópico, cinco (50%) lograron la
curación en seis meses, con un tiempo medio de curación de 161 días [ 7 ].
Otros inhibidores de la calcineurina podrían ser
beneficiosos para la PG, pero hay menos datos disponibles sobre estos agentes.
Un informe de caso documentó una mejoría con crema de pimecrolimus al 1% [ 17
]. Se ha descrito mejoría con ciclosporina tópica o intralesional en algunos
pacientes [ 18-21 ].
Dapsona oral
Administración y precauciones: Nuestra dosis habitual
de dapsona oscila entre 100 y 150 mg al día. Normalmente, iniciamos el
tratamiento en adultos con 50 mg al día y aumentamos la dosis a 100 mg al día
después de 7 a 10 días (si las pruebas de laboratorio no detectan anomalías
significativas).
Los signos de mejoría suelen aparecer entre dos y
cuatro semanas después de la dosis óptima. Si la respuesta a la dosis de 100 mg
al día es deficiente y el paciente tolera la dapsona , consideramos aumentar la
dosis a 150 mg al día. Si no se observa respuesta en cuatro semanas con esta dosis,
suspendemos la dapsona. Los eventos adversos hematológicos pueden ser un factor
limitante para aumentar la dosis de dapsona.
Entre los efectos adversos de la dapsona se incluyen
hemólisis, agranulocitosis, metahemoglobinemia, síndrome de hipersensibilidad a
la dapsona y neuropatía periférica. Se requiere monitorización de laboratorio
para detectar toxicidad hematológica y hepática.
Los pacientes con deficiencia de glucosa-6-fosfato
deshidrogenasa (G6PD) presentan un mayor riesgo de toxicidad hematológica. El
cribado de la deficiencia de G6PD debe preceder al tratamiento con dapsona .
Eficacia – En informes de casos y series pequeñas, la
dapsona se ha asociado con mejoras en la PG cuando se administra como
monoterapia o como agente ahorrador de glucocorticoides [ 9,22-29 ]. En un
estudio retrospectivo de 27 pacientes que recibieron dapsona durante al menos
cuatro semanas como componente de la terapia con PG, se documentó una respuesta
completa o parcial para el 16 y el 81 por ciento de los pacientes,
respectivamente, con un tiempo promedio hasta la respuesta inicial de
aproximadamente cinco semanas [ 29 ].
Minociclina oral
Administración y precauciones: La dosis habitual de
minociclina oral para adultos es de 100 mg dos veces al día. Algunos médicos
utilizan doxiciclina como alternativa a la minociclina debido a su perfil de
efectos adversos más favorable; sin embargo, la mayoría de los informes
publicados sobre beneficios se refieren al uso de minociclina [ 22 ]. Si no se
observa respuesta en un plazo de seis a ocho semanas, se suspende la
minociclina.
Entre los posibles efectos adversos de la minociclina
se incluyen mareos, decoloración de la piel, fotosensibilidad y un síndrome
similar al lupus. Al igual que con otras tetraciclinas, no debe administrarse a
embarazadas ni a niños menores de nueve años debido a sus efectos adversos
sobre el desarrollo dental.
Eficacia: Los datos de eficacia de la minociclina son
limitados. Los informes de casos describen una mejoría en la PG cuando se
utiliza como monoterapia o como agente ahorrador de glucocorticoides [ 22,30-39
].
Enfermedad limitada refractaria: cuando la PG limitada no responde
adecuadamente a los corticosteroides tópicos, los inhibidores de la
calcineurina tópicos, la dapsona oral o la minociclina oral , procedemos a las
intervenciones utilizadas para la enfermedad más extensa.
Enfermedad más extensa y enfermedad limitada
resistente al tratamiento: Adoptamos un
enfoque diferente para el tratamiento de la PG que va más allá de nuestra
descripción de enfermedad limitada (es decir, más que unas pocas úlceras
superficiales pequeñas [p. ej., <3 cm] o una placa solitaria de PG
vegetativa). Esta extensión de la PG generalmente requiere terapia sistémica
para calmar el proceso inflamatorio y permitir la cicatrización de la herida.
La terapia sistémica también es apropiada para la PG limitada que no responde
adecuadamente a otros tratamientos.
Tratamiento inicial:
Nuestro tratamiento inicial preferido para la PG extensa es un
glucocorticoide sistémico o ciclosporina oral debido a la rápida aparición de
sus efectos antiinflamatorios. Seleccionamos el fármaco más tolerable según los
factores específicos del paciente, como las comorbilidades. Generalmente,
comenzamos con un glucocorticoide sistémico, reservando la ciclosporina para
pacientes que no toleran la terapia con glucocorticoides sistémicos.
Dado que el tratamiento a largo plazo con estas terapias suele asociarse con efectos adversos graves, solemos incorporar otras terapias inmunosupresoras sistémicas durante las primeras semanas de tratamiento.
El cuidado de las heridas y el manejo del dolor son
componentes importantes del tratamiento.
Los corticosteroides tópicos y el tacrolimus tópico se utilizan a menudo como complemento del tratamiento sistémico.
Glucocorticoides sistémicos
Administración y precauciones: Nuestro régimen
inicial habitual para adultos es de 0,5 a 1,5 mg/kg al día de prednisona oral o
su equivalente, con una dosis máxima de 60 mg de prednisona al día. En casos
muy agresivos o dolorosos, se pueden utilizar pulsos de corticosteroides
intravenosos ( metilprednisolona , 1 g al día durante uno a cinco días) como
tratamiento inicial [ 40-42 ]. Posteriormente, se cambia a un corticosteroide
oral.
La respuesta a los glucocorticoides sistémicos suele
ser rápida; la estabilización de la enfermedad suele ser evidente durante la
primera semana [ 43 ]. El dolor también tiende a mejorar poco después del
inicio del tratamiento.
Nuestro objetivo es reducir gradualmente la dosis de
glucocorticoides lo antes posible debido a la posibilidad de efectos adversos
graves con el tratamiento sistémico a largo plazo. En la mayoría de los
pacientes, esto nos lleva a añadir un agente sistémico adicional durante las primeras
semanas de tratamiento. (Véase "Manejo posterior" más adelante).
El tratamiento sistémico con glucocorticoides se
asocia a una amplia variedad de posibles efectos adversos. Este tratamiento
debe incluir medidas para reducir la pérdida ósea asociada a los
glucocorticoides.
Eficacia – Aunque múltiples informes de casos, series
de casos y nuestra experiencia clínica respaldan el uso de glucocorticoides
sistémicos para la PG [ 6,22 ], los hallazgos de un ensayo aleatorizado simple
ciego (n = 112) que comparó la monoterapia con prednisolona (dosis inicial de
0,75 mg/kg por día, hasta 75 mg por día) con ciclosporina 4 mg/kg por día
(hasta 400 mg por día) mostraron que la terapia con glucocorticoides sistémicos
por sí sola puede ser insuficiente para muchos pacientes [ 44 ]. Después de
seis meses, solo hubo una tasa del 47 por ciento de curación completa de la
úlcera en ambos grupos. El ritmo de mejoría durante las primeras seis semanas
de tratamiento fue similar entre los grupos, lo que sugiere un tiempo
comparable hasta el inicio de la acción. Las tasas de reacciones adversas
también fueron similares en ambos grupos, pero las reacciones graves, en
particular las infecciones graves, fueron más frecuentes en el grupo de
prednisolona.
Ciclosporina sistémica
Administración y precauciones: Nuestra pauta inicial
habitual para adultos es de 4 a 5 mg/kg de ciclosporina al día [ 45 ]. Al igual
que con los glucocorticoides orales, la respuesta suele ser rápida. El dolor
suele mejorar en pocos días y se esperan otros signos de mejoría durante las
primeras cuatro semanas.
Nuestro objetivo es reducir gradualmente la dosis de
ciclosporina lo antes posible debido a la preocupación por los efectos
adversos. En la mayoría de los pacientes, esto nos lleva a añadir un agente
sistémico adicional durante las primeras semanas de tratamiento.
Entre los efectos adversos de la ciclosporina se
incluyen la toxicidad renal, la hipertensión y el aumento del riesgo de
infecciones y neoplasias malignas. Los efectos adversos se analizan con más
detalle por separado. Se recomienda la monitorización periódica de la función
renal y la presión arterial. Las contraindicaciones de la ciclosporina incluyen
neoplasias malignas concurrentes, hipertensión no controlada e infecciones no
controladas.
Eficacia: Un ensayo aleatorizado simple ciego (n =
112) que comparó ciclosporina con prednisolona encontró curación completa
después de seis meses en aproximadamente la mitad de los pacientes tratados con
ciclosporina y un tiempo similar hasta el inicio de la acción entre
ciclosporina y prednisolona [ 44 ].
Además, varios análisis retrospectivos e informes de
casos sugieren un efecto beneficioso de la ciclosporina [ 9,22-24,44,46,47 ].
Gestión posterior
Rol e implementación:
En nuestra experiencia, la mayoría de los pacientes con PG extensa
requieren varios meses de tratamiento sistémico para lograr la curación. Las
terapias sistémicas distintas de los glucocorticoides y la ciclosporina (p.
ej., infliximab , adalimumab , micofenolato de mofetilo) pueden desempeñar un
papel importante en la curación de la PG y en la reducción del riesgo de
efectos adversos relacionados con el tratamiento sistémico a largo plazo con
glucocorticoides o ciclosporina.
Momento oportuno: Muchas de las terapias
inmunosupresoras preferidas para el tratamiento a largo plazo tienen un inicio
de acción más lento que los glucocorticoides sistémicos y la ciclosporina . Por
lo tanto, solemos añadir estas terapias de forma temprana, generalmente durante
las primeras semanas del tratamiento con glucocorticoides sistémicos o
ciclosporina. Esto facilita la reducción gradual de la dosis de
glucocorticoides y ciclosporina en un plazo razonable.
Una excepción ocasional es el paciente que experimenta
una cicatrización muy rápida de la úlcera durante las primeras semanas de
tratamiento con glucocorticoides sistémicos o ciclosporina . Si parece
improbable la necesidad de un tratamiento sistémico prolongado (p. ej., más de
dos a tres meses para glucocorticoides orales o más de tres a seis meses para
ciclosporina), a menudo posponemos la adición de otro tratamiento sistémico.
Selección del tratamiento: infliximab , adalimumab o
micofenolato de mofetilo son nuestras terapias preferidas a largo plazo para el
PG debido a los datos que sugieren su eficacia. Sin embargo, otros factores
pueden influir en la selección del tratamiento, como las comorbilidades, la
preferencia del paciente o la disponibilidad del tratamiento. Por ejemplo,
infliximab o adalimumab pueden ser preferibles al micofenolato de mofetilo en
pacientes con enfermedad de Crohn o colitis ulcerosa debido al beneficio de los
inhibidores biológicos del factor de necrosis tumoral (TNF) para la enfermedad
inflamatoria intestinal y el PG, y las contraindicaciones para la
inmunosupresión pueden llevar a la selección de inmunoglobulina intravenosa
(IVIG) sobre otras terapias. En otros casos, un inhibidor biológico de la
interleucina (IL) 17 o la IL-23 puede ser preferible debido a su eficacia para una
comorbilidad.
Reducción gradual e interrupción de glucocorticoides
sistémicos y ciclosporina: En pacientes que reciben un glucocorticoide
sistémico (o ciclosporina ) junto con otro fármaco sistémico, generalmente
comenzamos a reducir gradualmente la dosis del glucocorticoide o la
ciclosporina una vez que la enfermedad se ha detenido y se observan signos
claros de mejoría (reducción del dolor, la inflamación visible y el tamaño de
la herida). En pacientes que reciben solo glucocorticoide sistémico o
ciclosporina, debido a la alta probabilidad de lograr una curación completa con
relativa rapidez, comenzamos a reducir gradualmente el fármaco tras lograr la
curación completa.
Nuestro objetivo es reducir gradualmente la dosis y
suspender los glucocorticoides en un plazo de 4 a 10 semanas, y monitorizar
estrechamente a los pacientes para asegurar una mejoría continua. Una reducción
demasiado rápida de los glucocorticoides puede precipitar brotes de la
enfermedad [ 43 ].
Generalmente, reducimos gradualmente la dosis de
ciclosporina durante un período que es el doble de la duración del tratamiento
con ciclosporina (por ejemplo, reducimos gradualmente durante cinco a seis
meses para pacientes con una duración de tratamiento con ciclosporina de tres
meses).
Infliximab
Administración y precauciones: Los regímenes de
tratamiento con infliximab y el tiempo de respuesta varían considerablemente
entre los informes de casos y estudios [ 48 ]. No se ha determinado el régimen
de tratamiento óptimo. Normalmente administramos una infusión intravenosa de 5
mg/kg de infliximab en las semanas 0, 2 y 6, seguida de infusiones cada seis a
ocho semanas [ 45 ]. En nuestra experiencia, los signos iniciales de respuesta
suelen ser evidentes entre las 8 y las 12 semanas.
Los posibles efectos adversos del infliximab incluyen
reacciones a la infusión, infecciones, enfermedad desmielinizante e
insuficiencia cardíaca. Los efectos secundarios de los inhibidores del TNF-alfa
se describen con más detalle por separado. (Véase "Inhibidores del factor
de necrosis tumoral alfa: Resumen de los efectos adversos" ).
Eficacia: El uso de infliximab , un anticuerpo
quimérico contra el TNF-alfa, en el PG está respaldado por un ensayo
aleatorizado controlado con placebo de 30 adultos con PG, muchos de los cuales
recibieron tratamiento concomitante con terapia tópica o sistémica (con mayor
frecuencia prednisolona oral ) [ 49 ]. Al inicio del ensayo, los pacientes
recibieron una infusión única de 5 mg/kg de infliximab (n = 13) o placebo (n =
17). Después de dos semanas, más pacientes en el grupo de infliximab
presentaron signos clínicos de mejoría (46 frente a 6%).
Los pacientes de ambos grupos que no mostraron mejoría
en la semana 2 recibieron infliximab de forma abierta (dosis única de 5 mg/kg).
En la semana 6, 20 de los 29 pacientes que recibieron infliximab (69%) habían
mejorado y 6 pacientes alcanzaron la remisión completa (21%). No se observaron
diferencias significativas en la respuesta al tratamiento entre los pacientes
con enfermedad inflamatoria intestinal (n = 18) y sin ella (n = 11). No se
realizó un seguimiento a largo plazo.
Además del ensayo aleatorizado, otros estudios
sugieren la eficacia de infliximab [ 50,51 ]. Como ejemplo, un estudio
retrospectivo multicéntrico de 13 pacientes con PG refractaria y enfermedad
inflamatoria intestinal tratados con 5 mg/kg de infliximab (1 a 24 infusiones
durante cero a cuatro años y en conjunción con otras terapias sistémicas)
encontró que los 13 pacientes lograron la curación completa. Los tiempos medios
hasta la respuesta inicial y la curación completa fueron 11 días (rango de 2 a
30 días) y 86 días (rango de 7 a 210 días), respectivamente [ 51 ]. Diez
pacientes requirieron infusiones periódicas cada 4 a 12 semanas para mantener
la respuesta clínica. Todos los pacientes que tomaban glucocorticoides
sistémicos pudieron suspender la terapia con glucocorticoides.
Adalimumab
Administración y precauciones: Al igual que con
infliximab , los regímenes de tratamiento con adalimumab varían [ 52-55 ].
Nuestro enfoque habitual comienza con una dosis de 40 mg de adalimumab cada dos
semanas. Los signos de mejoría suelen hacerse evidentes en un plazo de 8 a 12
semanas. En nuestra experiencia, a menudo se requiere un aumento posterior a
una dosis semanal para lograr la curación completa.
Los posibles efectos adversos incluyen reacciones en
el lugar de la inyección, infecciones, enfermedad desmielinizante e
insuficiencia cardíaca. Los efectos secundarios de los inhibidores del TNF-alfa
se describen con más detalle por separado. (Véase "Inhibidores del factor
de necrosis tumoral alfa: Resumen de los efectos adversos" ).
Eficacia: Un estudio abierto de 22 adultos con PG
tratados con adalimumab (dosis de 160 mg en la semana 0, seguidas de 80 mg en
la semana 2 y, posteriormente, 40 mg en semanas alternas a partir de la semana
4) sugiere un beneficio del adalimumab [ 56 ]. Dieciséis pacientes (73%)
también recibieron corticosteroides orales (≤10 mg al día) para PG u otras
indicaciones. En la semana 26, 12 pacientes lograron la cicatrización completa
de la úlcera diana (55%, IC del 95%: 32-76%). El tiempo medio hasta la
cicatrización de la úlcera diana fue de 115 días. El adalimumab también se ha
asociado con la cicatrización de la úlcera en informes de pacientes
individuales [ 52-55 ].
Micofenolato de mofetilo
Administración y precauciones : La dosis habitual de
micofenolato de mofetilo para el tratamiento de la PG en adultos es de 2 a 3 g
al día. Se espera una respuesta inicial en un plazo de 8 a 12 semanas.
Entre los efectos adversos del micofenolato de mofetilo se incluyen síntomas gastrointestinales y supresión de la médula ósea. Los efectos adversos se analizan con más detalle por separado.
Eficacia – Se han reportado múltiples casos de mejoría
en PG con micofenolato de mofetilo [ 9,24,57-62 ]. El micofenolato de mofetilo
pareció ser beneficioso en un estudio retrospectivo de 26 pacientes con PG
tratados con micofenolato de mofetilo y prednisolona (con o sin otros agentes
inmunomoduladores) durante al menos un mes [ 62 ]. Se observó mejoría en la
gravedad de la enfermedad durante el tratamiento en 22 pacientes (85%). Además,
en un estudio retrospectivo de siete pacientes tratados con micofenolato de
mofetilo durante al menos dos meses, cuatro de siete tuvieron curación
completa, incluidos dos pacientes para quienes el micofenolato de mofetilo fue
la única terapia sistémica administrada para PG [ 61 ].
Enfermedad refractaria: Cuando nuestro enfoque terapéutico habitual
resulta insuficiente, procedemos a otras terapias. Dado que los datos de
eficacia son limitados, la selección del tratamiento de siguiente línea se basa
principalmente en la consideración de las comorbilidades del paciente, sus
preferencias y la disponibilidad del tratamiento. (Véase "Otras
intervenciones" más adelante).
Suspensión del tratamiento: Una vez que las lesiones hayan cicatrizado
por completo, se puede intentar suspender el tratamiento. La terapia sistémica
debe reducirse gradualmente y suspenderse a lo largo de varios meses, en lugar
de suspenderse bruscamente.
Algunos pacientes requieren un tratamiento prolongado.
En una serie de 42 pacientes con PG, con un seguimiento medio de 26,5 meses, el
56 % de los 34 pacientes que sobrevivieron durante el seguimiento requirieron
terapia farmacológica continua, y el 44 % logró mantener la remisión completa
sin terapia [ 23 ].
Manejo de heridas
El objetivo del cuidado de heridas en PG es crear un
entorno óptimo para la cicatrización de la herida.
Cuidado local:
Las heridas deben limpiarse suavemente con solución salina estéril tibia
o un antiséptico suave antes de cambiar el apósito [ 2 ]. Se prefieren los
apósitos que promueven un ambiente húmedo y no se adhieren a la base de la
herida, ya que pueden ser beneficiosos para la cicatrización [ 63 ].
La selección de un tipo específico de apósito para
heridas depende de la naturaleza de la herida y de las preferencias del
paciente y del profesional sanitario. Por ejemplo, los pacientes con heridas
con alto grado de exudación pueden beneficiarse del uso de apósitos más
absorbentes, como los alginatos, para evitar la maceración tisular [ 64 ].
Además de la zona de ulceración o erosión, también se
debe prestar atención a la piel circundante. El uso de una crema o ungüento
protector, como pasta de óxido de zinc o vaselina, puede ayudar a prevenir la
rotura de la piel en el borde de la herida [ 63,64 ].
Los pacientes deben ser monitoreados para detectar
signos clínicos de infección de la herida (por ejemplo, fiebre, calor,
hinchazón, mal olor, estrías linfangíticas, aumento del drenaje o dolor) y
deben ser tratados adecuadamente con antibióticos si ocurre una infección [ 64
].
Prevención de traumatismos: La patergia (exacerbación o desarrollo de
nuevas lesiones en los sitios de traumatismo) puede ocurrir en el PG. Por lo
tanto, deben evitarse las lesiones traumáticas innecesarias en la herida, como
el uso de apósitos húmedos o secos y la aplicación de sustancias cáusticas (p.
ej., nitrato de plata ) [ 2,64 ].
Si un paciente con PG activo requiere cirugía por otra
indicación, recomendamos continuar el tratamiento con PG en la medida de lo
posible y suspender la reducción gradual de las terapias con PG durante el
período perioperatorio y hasta la cicatrización de la herida quirúrgica.
Idealmente, las intervenciones quirúrgicas electivas deben posponerse hasta la
cicatrización completa del PG.
Papel de la cirugía y otras intervenciones :
debido al potencial de patergia, el papel de la cirugía para el manejo
de heridas en PG es controvertido [ 65 ]. Los procedimientos quirúrgicos se
consideran solo en casos selectos, como aquellos en los que la acumulación de
tejido necrótico presenta un riesgo de infección o donde tejidos vitales como
tendones o ligamentos están expuestos en el lecho de la úlcera [ 6,45 ].
Aunque se han reportado múltiples casos en los que el
PG empeoró después de una intervención quirúrgica [ 1,66 ], existe
documentación de casos en los que el desbridamiento suave, los colgajos o
injertos de piel [ 67-70 ], los injertos de espesor parcial con terapia de
presión negativa para heridas [ 71 ] y la aplicación de autoinjertos de
queratinocitos bioingenierizados [ 58 ] o sustitutos dérmicos cultivados
alogénicos [ 72 ] parecieron ser beneficiosos en el PG. En una revisión de la
literatura que identificó casos de intervención quirúrgica en pacientes con PG
activo, se identificó progresión de la enfermedad postoperatoria en solo el 17
por ciento de los pacientes [ 65 ].
Si se va a realizar una cirugía para una úlcera por
PG, recomendamos posponerla hasta que la enfermedad se haya controlado
adecuadamente (es decir, hasta que se haya resuelto el eritema o la coloración
violácea en el borde de la herida y se haya detenido la progresión de la
enfermedad). Los pacientes deben recibir tratamiento concomitante con terapia
sistémica para PG [ 73 ].
Aunque el cierre del estoma puede conducir a la
resolución de la PG periostomal, su recurrencia puede ser frecuente tras la
reubicación o revisión del estoma. En una revisión retrospectiva de historias
clínicas de 44 pacientes con PG periostomal, 10 de 15 pacientes (67%) sometidos
a reubicación o revisión del estoma desarrollaron PG recurrente [ 74 ].
Se ha informado que el oxígeno hiperbárico es
beneficioso para la cicatrización de heridas en algunos pacientes con PG, pero
los datos son insuficientes para recomendar el uso rutinario de esta terapia [
75-77 ].
Manejo del dolor
OTRAS INTERVENCIONES
Se han utilizado otras intervenciones para la PG, pero
la limitada información sobre eficacia, los perfiles de efectos adversos
desfavorables, el coste o la disponibilidad generalmente restringen su uso a la
enfermedad refractaria y a situaciones en las que un tratamiento específico
puede ofrecer un beneficio conjunto para la PG y una comorbilidad. Ejemplos de
estas intervenciones incluyen otros agentes biológicos ( adalimumab ,
inhibidores de la interleucina [IL] 17, inhibidores de la IL-23), otros inmunosupresores
convencionales ( metotrexato , azatioprina ), inmunoglobulina intravenosa
(IGIV), agentes alquilantes y otros tratamientos.
Otros inhibidores biológicos del TNF: En nuestra
experiencia, infliximab y adalimumab son más eficaces que otros inhibidores del
factor de necrosis tumoral (TNF), aunque se han descrito beneficios con otros
inhibidores del TNF. También se ha descrito una mejoría en la PG con etanercept
(en dosis de 25 a 50 mg dos veces por semana) en una pequeña serie
retrospectiva y un informe de caso [ 78,79 ]. Existen informes de caso que
describen los beneficios de otros inhibidores biológicos del TNF-alfa, como
certolizumab pegol [ 80,81 ] y golimumab [ 82 ].
Inhibidores biológicos de IL-17 e IL-23: los informes
de casos, la mayoría describiendo la terapia biológica como un tratamiento
adyuvante para PG en pacientes con enfermedad inflamatoria intestinal
concomitante, sugieren beneficio de ustekinumab , un inhibidor
anti-interleucina (IL) 12/23 [ 83-85 ], así como los agentes biológicos
anti-IL-23 tildrakizumab [ 86 ], guselkumab [ 87 ] y risankizumab [ 88 ]. El
uso de biológicos anti-IL-17 para PG es incierto, con ambos casos de inducción
y depuración de PG en secukinumab [ 89,90 ], ixekizumab [ 91,92 ] y brodalumab
[ 93,94 ].
Los inmunosupresores convencionales – metotrexato [ 95
] y azatioprina [ 23,96 ] han resultado beneficiosos como tratamiento adyuvante
o ahorrador de glucocorticoides en informes de casos y series de casos pequeñas
[ 97 ]. De manera similar al micofenolato de mofetilo, estos fármacos tienen un
inicio de acción retardado (generalmente de 8 a 12 semanas).
La dosis habitual de metotrexato para adultos es de 10
a 30 mg semanales ; la azatioprina suele prescribirse en dosis de 100 a 300 mg
diarios [ 45 ]. La actividad de la enzima tiopurina metiltransferasa (TPMT)
determina la dosis adecuada de azatioprina. Ambos fármacos pueden causar
efectos adversos graves.
IgIV : La inmunoglobulina intravenosa (IGIV) se
administra generalmente en una dosis total de 2 g/kg cada cuatro semanas. La
dosis total se divide y se administra en un período de dos a cinco días.
Los datos sobre la eficacia de la IgIV para el PG son
limitados. Una revisión sistemática de casos y series de casos que documentan
el tratamiento del PG refractario con IgIV (a menudo junto con glucocorticoides
sistémicos) halló respuestas completas en 26 de 49 pacientes (53 %) y
respuestas completas o parciales en 43 de 49 pacientes (88 %) [ 98 ]. El tiempo
medio hasta la respuesta inicial fue de 3,5 semanas, y los pacientes recibieron
tratamiento durante un promedio de 5,9 meses. Las limitaciones de la revisión
incluyeron un posible sesgo de publicación, dado que la mayoría de los datos se
derivaron de informes de casos y la escasez de datos de seguimiento a largo
plazo de los pacientes incluidos. Además, se han observado casos de fracaso en
la respuesta al retratamiento con IgIV tras una recaída de la enfermedad. Se
necesitan ensayos prospectivos bien diseñados para aclarar la eficacia de la
IgIV para el PG. (Véase "Resumen de la terapia con inmunoglobulina
intravenosa (IgIV)" ).
Ciclofosfamida y clorambucilo: aunque algunos
informes de casos y estudios no controlados documentan una mejoría en la PG
refractaria con ciclofosfamida intravenosa pulsada [ 99-101 ] o clorambucilo [
102-104 ], la preocupación por la mielosupresión y otros efectos adversos
graves limita el uso de estos agentes alquilantes. En un estudio no controlado
de nueve pacientes con PG tratados con infusiones mensuales de ciclofosfamida
(a una dosis de 500 mg/m² durante hasta seis tratamientos), siete pacientes
lograron una remisión completa, uno logró una remisión parcial y uno no
respondió a la terapia [ 99 ]. Cuatro pacientes lograron la curación completa
antes del tercer tratamiento. Dos pacientes recayeron dentro de los tres meses
posteriores al cese de la terapia.
El clorambucilo pareció ser eficaz para el PG
recalcitrante en un estudio no controlado de seis pacientes tratados con
clorambucilo (en dosis de 2 a 4 mg al día) con o sin prednisona [ 102 ]. Se
observó una mejoría en seis a ocho semanas, y la cicatrización de las úlceras
se produjo en un plazo de 2 a 10 meses en todos los pacientes. Todos los
pacientes que también recibían prednisona para el PG pudieron suspender la
terapia con prednisona. Se mantuvieron remisiones prolongadas (de cuatro a
nueve años) en cuatro pacientes que suspendieron el clorambucilo después de 6 a
24 meses de tratamiento.
Otros agentes: Se ha informado que otras
intervenciones son beneficiosas en pacientes individuales. Ejemplos de terapias
tópicas incluyen cromoglicato de sodio [ 9,105,106 ], nicotina [ 107-109 ],
peróxido de benzoilo [ 110,111 ], ácido 5-aminosalicílico [ 112 ], mostaza
nitrogenada [ 113 ] y factor de crecimiento derivado de plaquetas [ 114,115 ].
Otros agentes biológicos con informes de beneficio
aparente incluyen el inhibidor de IL-1 anakinra [ 116 ], el inhibidor de
IL-1-beta canakinumab [ 117 ], el antagonista del receptor IL-36 spesolimab [
118 ] y el inhibidor de IL-6 tocilizumab [ 119 ]. Los inhibidores de la Janus
quinasa (JAK) con informes de beneficio aparente incluyen baricitinib [ 120,121
] y tofacitinib [ 122-125 ].
Los agentes sistémicos adicionales que pueden ser
beneficiosos incluyen clofazimina [ 23,126 ], colchicina [ 127,128 ],
interferón alfa [ 129 ], melfalán [ 130 ], mercaptopurina [ 131 ], metronidazol
[ 132 ], yoduro de potasio [ 133 ], sulfasalazina [ 24 ], tacrolimus [ 134 ] y
talidomida [ 22,135,136 ]. La aféresis de leucocitos [ 137-141 ] y la
plasmaféresis [ 101 ] también se han utilizado para el tratamiento.
PRONÓSTICO
Los estudios de seguimiento de pacientes con PG
sugieren que con tratamiento, más del 50 por ciento de los pacientes logran una
curación completa de la herida en el plazo de un año:
En una serie australiana de 26 pacientes
hospitalizados por PG, 6 de 12 pacientes disponibles para seguimiento seis
meses después del alta hospitalaria tuvieron curación completa de la úlcera [
142 ].
En una serie de 64 pacientes con PG clásica y 22
pacientes con PG ampollosa (atípica), el tiempo medio hasta la remisión
completa (cicatrización de la herida y ausencia de enfermedad activa) fue de
11,5 ± 11,1 meses en pacientes con PG clásica y de 9 ± 13,7 meses en pacientes
con PG ampollosa [ 1 ]. En general, el 68 por ciento de los pacientes alcanzó
la remisión completa en un plazo de seis meses y el 95 por ciento estuvo en
remisión en un plazo de tres años.
El efecto del tratamiento de trastornos médicos
subyacentes asociados sobre la actividad de la enfermedad en pacientes con PG
es variable, ya que el curso de la PG no siempre es paralelo al de la
enfermedad subyacente relacionada [ 9 ]. Se ha informado de una mejoría en la
PG en algunos pacientes después de una proctocolectomía por enfermedad
inflamatoria intestinal [ 143 ]. Sin embargo, la PG también puede ocurrir
periestomalmente o en otras ubicaciones después de una proctocolectomía [
9,144-148 ].
El PG suele curarse con cicatrices, que pueden ser
desfigurantes. Pueden desarrollarse nuevas lesiones durante o después de la
curación de otras lesiones. Las recaídas pueden aparecer tras largos periodos
de inactividad de la enfermedad [ 149 ].
La PG puede estar asociada a un mayor riesgo de
muerte. En un estudio de cohorte retrospectivo que incluyó a 3386 pacientes con
PG y más de 67 000 controles, la mortalidad por cualquier causa fue mayor en
pacientes con PG (cociente de riesgos instantáneos ajustado: 2,122; IC del 95
%: 1,971-2,285) [ 150 ]. También se observaron mayores tasas de mortalidad en
enfermedades de diversos sistemas orgánicos.
Diversos factores pueden contribuir al aumento de la
mortalidad en pacientes con PG, y la muerte puede ocurrir debido a trastornos
asociados subyacentes, particularmente en pacientes con malignidad, o debido a
complicaciones directamente relacionadas con úlceras o terapia inmunosupresora
(por ejemplo, sepsis) [ 23,151 ]. También se han propuesto otros posibles
factores contribuyentes, como los efectos relacionados con un estado
inflamatorio crónico subyacente [ 150 ].
Debido a la asociación entre la PG ampollosa y la
enfermedad hematológica, los pacientes con PG ampollosa que no presentan un
trastorno hematológico subyacente identificable deben ser seguidos de cerca [
45 ].
REFERENCIAS
- Ruocco E, Sangiuliano S, Gravina AG, et al. Pyoderma gangrenosum: an updated review. J Eur Acad Dermatol Venereol 2009; 23:1008.
- Allen CP, Hull J, Wilkison N, Burge SM. Pediatric pyoderma gangrenosum with splenic and pulmonary involvement. Pediatr Dermatol 2013; 30:497.
- Bhat RM, Shetty SS, Kamath GH. Pyoderma Gangrenosum in childhood. Int J Dermatol 2004; 43:205.
- Torrelo A, Colmenero I, Serrano C, et al. Pyoderma gangrenosum in an infant. Pediatr Dermatol 2006; 23:338.
- von den Driesch P. Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol 1997; 137:1000.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore) 2000; 79:37.
- Binus AM, Qureshi AA, Li VW, Winterfield LS. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol 2011; 165:1244.
- Saracino A, Kelly R, Liew D, Chong A. Pyoderma gangrenosum requiring inpatient management: a report of 26 cases with follow up. Australas J Dermatol 2011; 52:218.
- Marzano AV, Damiani G, Ceccherini I, et al. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol 2017; 176:1588.
- Ortega-Loayza AG, Nugent WH, Lucero OM, et al. Dysregulation of inflammatory gene expression in lesional and nonlesional skin of patients with pyoderma gangrenosum. Br J Dermatol 2018; 178:e35.
- Ortega-Loayza AG, Friedman MA, Reese AM, et al. Molecular and Cellular Characterization of Pyoderma Gangrenosum: Implications for the Use of Gene Expression. J Invest Dermatol 2022; 142:1217.
- Lu JD, Milakovic M, Ortega-Loayza AG, et al. Pyoderma gangrenosum: proposed pathogenesis and current use of biologics with an emphasis on complement C5a inhibitor IFX-1. Expert Opin Investig Drugs 2020; 29:1179.
- Alberts JH, Sams HH, Miller JL, King LE Jr. Familial ulcerative pyoderma gangrenosum: a report of 2 kindred. Cutis 2002; 69:427.
- Khandpur S, Mehta S, Reddy BS. Pyoderma gangrenosum in two siblings: a familial predisposition. Pediatr Dermatol 2001; 18:308.
- al-Rimawi HS, Abuekteish FM, Daoud AS, Oboosi MM. Familial pyoderma gangrenosum presenting in infancy. Eur J Pediatr 1996; 155:759.
- Shands JW Jr, Flowers FP, Hill HM, Smith JO. Pyoderma gangrenosum in a kindred. Precipitation by surgery or mild physical trauma. J Am Acad Dermatol 1987; 16:931.
- Yeon HB, Lindor NM, Seidman JG, Seidman CE. Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome maps to chromosome 15q. Am J Hum Genet 2000; 66:1443.
- Oprea Y, Antohi DR, Vague M, et al. Human Inborn Errors of Immunity in Pyoderma Gangrenosum: A Systematic Review. Am J Clin Dermatol 2024; 25:701.
- Smith KN, Welborn M, Monir RL, et al. Pediatric pyoderma gangrenosum associated with leukocyte adhesion deficiency type 1: A case report and review of the literature. Pediatr Dermatol 2023; 40:1086.
- Powell FC, Hackett BC, Wallach D. Pyoderma gangrenosum. In: Fitzpatrick's Dermatology in General Medicine, 8th ed, Goldsmith LA, Katz SI, Gilchrest BA, et al (Eds), McGraw-Hill Companies, Inc., 2012. Vol 1, p.371.
- Wong WW, Machado GR, Hill ME. Pyoderma gangrenosum: the great pretender and a challenging diagnosis. J Cutan Med Surg 2011; 15:322.
- Xia FD, Liu K, Lockwood S, et al. Risk of developing pyoderma gangrenosum after procedures in patients with a known history of pyoderma gangrenosum-A retrospective analysis. J Am Acad Dermatol 2018; 78:310.
- Callen JP, Jackson JM. Pyoderma gangrenosum: an update. Rheum Dis Clin North Am 2007; 33:787.
- Nico MM, Hussein TP, Aoki V, Lourenço SV. Pyostomatitis vegetans and its relation to inflammatory bowel disease, pyoderma gangrenosum, pyodermatitis vegetans, and pemphigus. J Oral Pathol Med 2012; 41:584.
- Brown TS, Marshall GS, Callen JP. Cavitating pulmonary infiltrate in an adolescent with pyoderma gangrenosum: a rarely recognized extracutaneous manifestation of a neutrophilic dermatosis. J Am Acad Dermatol 2000; 43:108.
- Fukuhara K, Urano Y, Kimura S, et al. Pyoderma gangrenosum with rheumatoid arthritis and pulmonary aseptic abscess responding to treatment with dapsone. Br J Dermatol 1998; 139:556.
- Abdelrazeq AS, Lund JN, Leveson SH. Pouchitis-associated pyoderma gangrenosum following restorative proctocolectomy for ulcerative colitis. Eur J Gastroenterol Hepatol 2004; 16:1057.
- Nurre LD, Rabalais GP, Callen JP. Neutrophilic dermatosis-associated sterile chronic multifocal osteomyelitis in pediatric patients: case report and review. Pediatr Dermatol 1999; 16:214.
- East-Innis A, Desnoes R, Thame K, et al. Pyoderma gangrenosum associated with osteomyelitis in a paediatric patient: a case report. West Indian Med J 2005; 54:207.
- Goldshmid O, Dovorish Z, Zehavi T, et al. Coexistent pyoderma gangrenosum and tibialis anterior myositis as presenting manifestations of Crohn's disease: case report and review of the literature. Rheumatol Int 2011; 31:525.
- Wilson DM, John GR, Callen JP. Peripheral ulcerative keratitis--an extracutaneous neutrophilic disorder: report of a patient with rheumatoid arthritis, pustular vasculitis, pyoderma gangrenosum, and Sweet's syndrome with an excellent response to cyclosporine therapy. J Am Acad Dermatol 1999; 40:331.
- Brown BA, Parker CT, Bower KS. Effective steroid-sparing treatment for peripheral ulcerative keratitis and pyoderma gangrenosum. Cornea 2001; 20:117.
- Teasley LA, Foster CS, Baltatzis S. Sclerokeratitis and facial skin lesions: a case report of pyoderma gangrenosum and its response to dapsone therapy. Cornea 2007; 26:215.
- Vadillo M, Jucgla A, Podzamczer D, et al. Pyoderma gangrenosum with liver, spleen and bone involvement in a patient with chronic myelomonocytic leukaemia. Br J Dermatol 1999; 141:541.
- Chanson P, Timsit J, Kujas M, et al. Pituitary granuloma and pyoderma gangrenosum. J Endocrinol Invest 1990; 13:677.
- Koester G, Tarnower A, Levisohn D, Burgdorf W. Bullous pyoderma gangrenosum. J Am Acad Dermatol 1993; 29:875.
- Scherlinger M, Guillet S, Doutre MS, et al. Pyoderma gangrenosum with extensive pulmonary involvement. J Eur Acad Dermatol Venereol 2017; 31:e214.
- Tolkachjov SN, Fahy AS, Wetter DA, et al. Postoperative pyoderma gangrenosum (PG): the Mayo Clinic experience of 20 years from 1994 through 2014. J Am Acad Dermatol 2015; 73:615.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The Association of Age With Clinical Presentation and Comorbidities of Pyoderma Gangrenosum. JAMA Dermatol 2018; 154:409.
- Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol 2012; 13:191.
- Marzano AV, Trevisan V, Gattorno M, et al. Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (PAPASH): a new autoinflammatory syndrome associated with a novel mutation of the PSTPIP1 gene. JAMA Dermatol 2013; 149:762.
- Braun-Falco M, Kovnerystyy O, Lohse P, Ruzicka T. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)--a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol 2012; 66:409.
- Marzano AV, Ishak RS, Colombo A, et al. Pyoderma gangrenosum, acne and suppurative hidradenitis syndrome following bowel bypass surgery. Dermatology 2012; 225:215.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore) 2014; 93:e187.
- Koshelev MV, Garrison PA, Wright TS. Concurrent hidradenitis suppurativa, inflammatory acne, dissecting cellulitis of the scalp, and pyoderma gangrenosum in a 16-year-old boy. Pediatr Dermatol 2014; 31:e20.
- Croitoru DO, Huang RS, McMullen EP, et al. Investigating historic cases of pyoderma gangrenosum in myelodysplastic syndrome and chronic myelomonocytic leukemia for possible VEXAS syndrome: A systematic review. J Am Acad Dermatol 2024; 91:712.
- Al Ghazal P, Herberger K, Schaller J, et al. Associated factors and comorbidities in patients with pyoderma gangrenosum in Germany: a retrospective multicentric analysis in 259 patients. Orphanet J Rare Dis 2013; 8:136.
- Jeong HS, Layher H, Cao L, et al. Pyoderma gangrenosum (PG) associated with levamisole-adulterated cocaine: Clinical, serologic, and histopathologic findings in a cohort of patients. J Am Acad Dermatol 2016; 74:892.
- Croitoru D, Nathanielsz N, Seigel K, et al. Clinical manifestations and treatment outcomes of pyoderma gangrenosum following rituximab exposure: A systematic review. J Am Acad Dermatol 2022; 87:655.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol 2004; 43:790.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic Criteria of Ulcerative Pyoderma Gangrenosum: A Delphi Consensus of International Experts. JAMA Dermatol 2018; 154:461.
- Jockenhöfer F, Wollina U, Salva KA, et al. The PARACELSUS score: a novel diagnostic tool for pyoderma gangrenosum. Br J Dermatol 2019; 180:615.
- Weedon D. The vasculopathic reaction pattern. In: Weedon's Skin Pathology, 3rd ed, Elsevier Limited, 2010. p.195.
- Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med 2002; 347:1412.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical Features of Neutrophilic Dermatosis Variants Resembling Necrotizing Fasciitis. JAMA Dermatol 2019; 155:79.
- Touil LL, Gurusinghe DA, Sadri A, et al. Postsurgical Pyoderma Gangrenosum Versus Necrotizing Fasciitis: Can We Spot the Difference? Ann Plast Surg 2017; 78:582.
- Hradil E, Jeppsson C, Hamnerius N, Svensson Å. The diagnosis you wish you had never operated on: Pyoderma gangrenosum misdiagnosed as necrotizing fasciitis-a case report. Acta Orthop 2017; 88:231.
- Berger N, Ebenhoch M, Salzmann M. Postoperative Pyoderma Gangrenosum in Children: The Case Report of a 13-Year-Old Boy With Pyoderma Gangrenosum After Hip Reconstruction Surgery and a Review of the Literature. J Pediatr Orthop 2017; 37:e379.
REFERENCIAS DE TRATAMIENTO Y PRONÓSTICO
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore) 2000; 79:37.
- Powell FC, Hackett BC, Wallach D. Pyoderma gangrenosum. In: Fitzpatrick's Dermatology in General Medicine, 8th ed, Goldsmith LA, Katz SI, Gilchrest BA, et al (Eds), McGraw-Hill Companies, Inc., 2012. Vol 1, p.371.
- Maronese CA, Pimentel MA, Li MM, et al. Pyoderma Gangrenosum: An Updated Literature Review on Established and Emerging Pharmacological Treatments. Am J Clin Dermatol 2022; 23:615.
- Prajapati V, Man J, Brassard A. Pyoderma gangrenosum: common pitfalls in management and a stepwise, evidence-based, therapeutic approach. J Cutan Med Surg 2009; 13 Suppl 1:S2.
- Le Cleach L, Moguelet P, Perrin P, Chosidow O. Is topical monotherapy effective for localized pyoderma gangrenosum? Arch Dermatol 2011; 147:101.
- Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol 2005; 53:273.
- Thomas KS, Ormerod AD, Craig FE, et al. Clinical outcomes and response of patients applying topical therapy for pyoderma gangrenosum: A prospective cohort study. J Am Acad Dermatol 2016; 75:940.
- Lyon CC, Stapleton M, Smith AJ, et al. Topical tacrolimus in the management of peristomal pyoderma gangrenosum. J Dermatolog Treat 2001; 12:13.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA 2000; 284:1546.
- Dohan FC. Schizophrenia and neuroactive peptides from food. Lancet 1979; 1:1031.
- Jennings JL. Pyoderma gangrenosum: successful treatment with intralesional steroids. J Am Acad Dermatol 1983; 9:575.
- Moschella SL. Pyoderma gangrenosum. A patient successfully treated with intralesional injections of steroid. Arch Dermatol 1967; 95:121.
- Altieri M, Vaziri K, Orkin BA. Topical tacrolimus for parastomal pyoderma gangrenosum: a report of two cases. Ostomy Wound Manage 2010; 56:56.
- Marzano AV, Trevisan V, Lazzari R, Crosti C. Topical tacrolimus for the treatment of localized, idiopathic, newly diagnosed pyoderma gangrenosum. J Dermatolog Treat 2010; 21:140.
- Kimble RM, Tickler AK, Nicholls VS, Cleghorn G. Successful topical tacrolimus (FK506) therapy in a child with pyoderma gangrenosum. J Pediatr Gastroenterol Nutr 2002; 34:555.
- Larsen CG, Thyssen JP. Pustular penile pyoderma gangrenosum successfully treated with topical tacrolimus ointment. Acta Derm Venereol 2012; 92:104.
- Bellini V, Simonetti S, Lisi P. Successful treatment of severe pyoderma gangrenosum with pimecrolimus cream 1%. J Eur Acad Dermatol Venereol 2008; 22:113.
- Mrowietz U, Christophers E. Clearing of pyoderma gangrenosum by intralesional cyclosporin A. Br J Dermatol 1991; 125:499.
- Wenzel J, Gerdsen R, Phillipp-Dormston W, et al. Topical treatment of pyoderma gangraenosum. Dermatology 2002; 205:221.
- Theissen U, Luger TA, Schwarz T. [Successful topical administration of cyclosporin A in pyoderma gangraenosum]. Hautarzt 1996; 47:132.
- Azizan NZ, Gangaram HB, Hussein SH. A novel therapy for the treatment of pyoderma gangrenosum. Med J Malaysia 2008; 63:51.
- Binus AM, Qureshi AA, Li VW, Winterfield LS. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol 2011; 165:1244.
- von den Driesch P. Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol 1997; 137:1000.
- Vidal D, Puig L, Gilaberte M, Alomar A. Review of 26 cases of classical pyoderma gangrenosum: clinical and therapeutic features. J Dermatolog Treat 2004; 15:146.
- Fukuhara K, Urano Y, Kimura S, et al. Pyoderma gangrenosum with rheumatoid arthritis and pulmonary aseptic abscess responding to treatment with dapsone. Br J Dermatol 1998; 139:556.
- Teasley LA, Foster CS, Baltatzis S. Sclerokeratitis and facial skin lesions: a case report of pyoderma gangrenosum and its response to dapsone therapy. Cornea 2007; 26:215.
- Brown RE, Lay L, Graham D. Bilateral pyoderma gangrenosum of the hand: treatment with dapsone. J Hand Surg Br 1993; 18:119.
- Galun E, Flugelman MY, Rachmilewitz D. Pyoderma gangrenosum complicating ulcerative colitis: successful treatment with methylprednisolone pulse therapy and dapsone. Am J Gastroenterol 1986; 81:988.
- Din RS, Tsiaras WG, Li DG, Mostaghimi A. Efficacy of Systemic Dapsone Treatment for Pyoderma Gangrenosum: A Retrospective Review. J Drugs Dermatol 2018; 17:1058.
- Farrell AM, Black MM, Bracka A, Bunker CB. Pyoderma gangrenosum of the penis. Br J Dermatol 1998; 138:337.
- Shenefelt PD. Pyoderma gangrenosum associated with cystic acne and hidradenitis suppurativa controlled by adding minocycline and sulfasalazine to the treatment regimen. Cutis 1996; 57:315.
- Boulinguez S, Bernard P, Bedane C, et al. Pyoderma gangrenosum complicating Cogan's syndrome. Clin Exp Dermatol 1998; 23:286.
- Langan SM, Powell FC. Vegetative pyoderma gangrenosum: a report of two new cases and a review of the literature. Int J Dermatol 2005; 44:623.
- Vandevyvere K, Luyten FP, Verschueren P, et al. Pyoderma gangrenosum developing during therapy with TNF-alpha antagonists in a patient with rheumatoid arthritis. Clin Rheumatol 2007; 26:2205.
- Sachs D, Su L, Dlugosz A. Verrucous annular ulcerated hip plaques. Diagnosis: superficial granulomatous pyoderma form of pyoderma gangrenosum. Arch Dermatol 2000; 136:1263.
- Miralles ES, Nuñez M, Pérez B, Ledo A. Minocycline hydrochloride hyperpigmentation complicating treatment of pyoderma gangrenosum. J Dermatol 1994; 21:965.
- Reynolds NJ, Peachey RD. Response of atypical bullous pyoderma gangrenosum to oral minocycline hydrochloride and topical steroids. Acta Derm Venereol 1990; 70:538.
- Davies MG, Piper S. Pyoderma gangrenosum: successful treatment with minocycline. Clin Exp Dermatol 1981; 6:219.
- Lynch WS, Bergfeld WF. Pyoderma gangrenosum responsive to minocycline hydrochloride. Cutis 1978; 21:535.
- Prystowsky JH, Kahn SN, Lazarus GS. Present status of pyoderma gangrenosum. Review of 21 cases. Arch Dermatol 1989; 125:57.
- Yamauchi T, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother 2003; 9:268.
- Aseni P, Di Sandro S, Mihaylov P, et al. Atypical presentation of pioderma gangrenosum complicating ulcerative colitis: rapid disappearance with methylprednisolone. World J Gastroenterol 2008; 14:5471.
- Chow RK, Ho VC. Treatment of pyoderma gangrenosum. J Am Acad Dermatol 1996; 34:1047.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ 2015; 350:h2958.
- Callen JP, Jackson JM. Pyoderma gangrenosum: an update. Rheum Dis Clin North Am 2007; 33:787.
- Hasselmann DO, Bens G, Tilgen W, Reichrath J. Pyoderma gangrenosum: clinical presentation and outcome in 18 cases and review of the literature. J Dtsch Dermatol Ges 2007; 5:560.
- Turner RB, Emer JJ, Weill M, et al. Rapid resolution of pyoderma gangrenosum after treatment with intravenous cyclosporine. J Am Acad Dermatol 2010; 63:e72.
- Adişen E, Oztaş M, Gürer MA. Treatment of idiopathic pyoderma gangrenosum with infliximab: induction dosing regimen or on-demand therapy? Dermatology 2008; 216:163.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut 2006; 55:505.
- Mooij JE, van Rappard DC, Mekkes JR. Six patients with pyoderma gangrenosum successfully treated with infliximab. Int J Dermatol 2013; 52:1418.
- Regueiro M, Valentine J, Plevy S, et al. Infliximab for treatment of pyoderma gangrenosum associated with inflammatory bowel disease. Am J Gastroenterol 2003; 98:1821.
- Hubbard VG, Friedmann AC, Goldsmith P. Systemic pyoderma gangrenosum responding to infliximab and adalimumab. Br J Dermatol 2005; 152:1059.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn's disease. Inflamm Bowel Dis 2009; 15:803.
- Reddick CL, Singh MN, Chalmers RJ. Successful treatment of superficial pyoderma gangrenosum associated with hidradenitis suppurativa with adalimumab. Dermatol Online J 2010; 16:15.
- Cariñanos I, Acosta MB, Domènech E. Adalimumab for pyoderma gangrenosum associated with inflammatory bowel disease. Inflamm Bowel Dis 2011; 17:E153.
- Yamasaki K, Yamanaka K, Zhao Y, et al. Adalimumab in Japanese patients with active ulcers of pyoderma gangrenosum: Twenty-six-week phase 3 open-label study. J Dermatol 2020; 47:1383.
- Nousari HC, Lynch W, Anhalt GJ, Petri M. The effectiveness of mycophenolate mofetil in refractory pyoderma gangrenosum. Arch Dermatol 1998; 134:1509.
- Wollina U, Karamfilov T. Treatment of recalcitrant ulcers in pyoderma gangrenosum with mycophenolate mofetil and autologous keratinocyte transplantation on a hyaluronic acid matrix. J Eur Acad Dermatol Venereol 2000; 14:187.
- Gilmour E, Stewart DG. Severe recalcitrant pyoderma gangrenosum responding to a combination of mycophenolate mofetil with cyclosporin and complicated by a mononeuritis. Br J Dermatol 2001; 144:397.
- Daniels NH, Callen JP. Mycophenolate mofetil is an effective treatment for peristomal pyoderma gangrenosum. Arch Dermatol 2004; 140:1427.
- Eaton PA, Callen JP. Mycophenolate mofetil as therapy for pyoderma gangrenosum. Arch Dermatol 2009; 145:781.
- Li J, Kelly R. Treatment of pyoderma gangrenosum with mycophenolate mofetil as a steroid-sparing agent. J Am Acad Dermatol 2013; 69:565.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol 2010; 62:646.
- Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol 2012; 13:191.
- Haag CK, Bacik L, Latour E, et al. Perioperative management of pyoderma gangrenosum. J Am Acad Dermatol 2020; 83:369.
- Barańska-Rybak W, Kakol M, Naesstrom M, et al. A retrospective study of 12 cases of pyoderma gangrenosum: why we should avoid surgical intervention and what therapy to apply. Am Surg 2011; 77:1644.
- Kim DW, Lee BI, Park SH. Accelerated healing of pyoderma gangrenosum in Behçet patient treated with cyclosporine and split thickness skin graft. Ann Plast Surg 2008; 61:552.
- Classen DA, Thomson C. Free flap coverage of pyoderma gangrenosum leg ulcers. J Cutan Med Surg 2002; 6:327.
- Zakhireh M, Rockwell WB, Fryer RH. Stabilization of pyoderma gangrenosum ulcer with oral cyclosporine prior to skin grafting. Plast Reconstr Surg 2004; 113:1417.
- Rozen SM, Nahabedian MY, Manson PN. Management strategies for pyoderma gangrenosum: case studies and review of literature. Ann Plast Surg 2001; 47:310.
- Pichler M, Larcher L, Holzer M, et al. Surgical treatment of pyoderma gangrenosum with negative pressure wound therapy and split thickness skin grafting under adequate immunosuppression is a valuable treatment option: Case series of 15 patients. J Am Acad Dermatol 2016; 74:760.
- Toyozawa S, Yamamoto Y, Nishide T, et al. Case report: a case of pyoderma gangrenosum with intractable leg ulcers treated by allogeneic cultured dermal substitutes. Dermatol Online J 2008; 14:17.
- Alam M, Grossman ME, Schneiderman PI, et al. Surgical management of pyoderma gangrenosum: case report and review. Dermatol Surg 2000; 26:1063.
- Barbosa NS, Tolkachjov SN, El-Azhary RA, et al. Clinical features, causes, treatments, and outcomes of peristomal pyoderma gangrenosum (PPG) in 44 patients: The Mayo Clinic experience, 1996 through 2013. J Am Acad Dermatol 2016; 75:931.
- Thomas CY Jr, Crouch JA, Guastello J. Hyperbaric oxygen therapy for pyoderma gangrenosum. Arch Dermatol 1974; 110:445.
- Davis JC, Landeen JM, Levine RA. Pyoderma gangrenosum: skin grafting after preparation with hyperbaric oxygen. Plast Reconstr Surg 1987; 79:200.
- Mazokopakis EE, Kofteridis DP, Pateromihelaki AT, et al. Improvement of ulcerative pyoderma gangrenosum with hyperbaric oxygen therapy. Dermatol Ther 2011; 24:134.
- Charles CA, Leon A, Banta MR, Kirsner RS. Etanercept for the treatment of refractory pyoderma gangrenosum: a brief series. Int J Dermatol 2007; 46:1095.
- Roy DB, Conte ET, Cohen DJ. The treatment of pyoderma gangrenosum using etanercept. J Am Acad Dermatol 2006; 54:S128.
- Pender TM, Ayandibu G, Van Voorhees AS. Certolizumab for the treatment of localized pyoderma gangrenosum associated with Crohn's disease: A case report. Dermatol Ther 2020; 33:e14352.
- Cinotti E, Labeille B, Perrot JL, et al. Certolizumab for the treatment of refractory disseminated pyoderma gangrenosum associated with rheumatoid arthritis. Clin Exp Dermatol 2014; 39:750.
- Diotallevi F, Campanati A, Radi G, et al. Pyoderma gangrenosum successfully treated with golimumab: Case report and review of the literature. Dermatol Ther 2019; 32:e12928.
- Guenova E, Teske A, Fehrenbacher B, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol 2011; 147:1203.
- Fahmy M, Ramamoorthy S, Hata T, Sandborn WJ. Ustekinumab for peristomal pyoderma gangrenosum. Am J Gastroenterol 2012; 107:794.
- Phillips FM, Verstockt B, Sebastian S, et al. Inflammatory Cutaneous Lesions in Inflammatory Bowel Disease Treated With Vedolizumab or Ustekinumab: An ECCO CONFER Multicentre Case Series. J Crohns Colitis 2020; 14:1488.
- John JM, Sinclair RD. Tildrakizumab for treatment of refractory pyoderma gangrenosum of the penis and polymyalgia rheumatica: Killing two birds with one stone. Australas J Dermatol 2020; 61:170.
- Reese AM, Erickson K, Reed KB, Ortega-Loayza AG. Modified dose of guselkumab for treatment of pyoderma gangrenosum. JAAD Case Rep 2022; 21:38.
- Burgdorf B, Schlott S, Ivanov IH, Dissemond J. Successful treatment of a refractory pyoderma gangrenosum with risankizumab. Int Wound J 2020; 17:1086.
- Orita A, Hoshina D, Hirosaki K. Pyoderma gangrenosum caused by secukinumab successfully treated with risankizumab: a case report and literature review. Clin Exp Dermatol 2022; 47:1372.
- Moreno García M, Madrid González M, Prada Lobato JM. Secukinumab for pyoderma gangrenosum: A case report. Med Clin (Barc) 2019; 152:246.
- Pollack IR, Wolner ZJ, Hammett J, Swerlick RA. Pyoderma gangrenosum in a patient on ixekizumab. JAAD Case Rep 2021; 16:152.
- Kao AS, King AD, Daveluy S. Successful treatment of cabozantinib-induced pyoderma gangrenosum with ixekizumab therapy: A case report. Dermatol Ther 2022; 35:e15716.
- Sadik CD, Thieme M, Zillikens D, Terheyden P. First emergence of pyoderma gangraenosum, palmoplantar pustulosis and sacroiliitis in a psoriasis patient associated with switching from secukinumab to brodalumab. J Eur Acad Dermatol Venereol 2019; 33:e406.
- Tee MW, Avarbock AB, Ungar J, Frew JW. Rapid resolution of pyoderma gangrenosum with brodalumab therapy. JAAD Case Rep 2020; 6:1167.
- Teitel AD. Treatment of pyoderma gangrenosum with methotrexate. Cutis 1996; 57:326.
- Sardar P, Guha P, Das NK, et al. Ulcerative pyoderma gangrenosum in mixed connective tissue disorder: a rare association and role of azathioprine in the management. Indian J Dermatol 2011; 56:600.
- Schmidt C, Wittig BM, Moser C, et al. Cyclophosphamide pulse therapy followed by azathioprine or methotrexate induces long-term remission in patients with steroid-refractory Crohn's disease. Aliment Pharmacol Ther 2006; 24:343.
- Song H, Lahood N, Mostaghimi A. Intravenous immunoglobulin as adjunct therapy for refractory pyoderma gangrenosum: systematic review of cases and case series. Br J Dermatol 2018; 178:363.
- Reynoso-von Drateln C, Perla-Navarro AV, Gamez-Nava JI, et al. Intravenous cyclophosphamide pulses in pyoderma gangrenosum: an open trial. J Rheumatol 1997; 24:689.
- Zonana-Nacach A, Jiménez-Balderas FJ, Martínez-Osuna P, Mintz G. Intravenous cyclophosphamide pulses in the treatment of pyoderma gangrenosum associated with rheumatoid arthritis: report of 2 cases and review of the literature. J Rheumatol 1994; 21:1352.
- Kaminska R, Ikäheimo R, Hollmen A. Plasmapheresis and cyclophosphamide as successful treatments for pyoderma gangrenosum. Clin Exp Dermatol 1999; 24:81.
- Burruss JB, Farmer ER, Callen JP. Chlorambucil is an effective corticosteroid-sparing agent for recalcitrant pyoderma gangrenosum. J Am Acad Dermatol 1996; 35:720.
- Callen JP, Case JD, Sager D. Chlorambucil--an effective corticosteroid-sparing therapy for pyoderma gangrenosum. J Am Acad Dermatol 1989; 21:515.
- Resnik BI, Rendon M, Kerdel FA. Successful treatment of aggressive pyoderma gangrenosum with pulse steroids and chlorambucil. J Am Acad Dermatol 1992; 27:635.
- de Cock KM, Thorne MG. The treatment of pyoderma gangrenosum with sodium cromoglycate. Br J Dermatol 1980; 102:231.
- Tamir A, Landau M, Brenner S. Topical treatment with 1% sodium cromoglycate in pyoderma gangrenosum. Dermatology 1996; 192:252.
- Patel GK, Rhodes JR, Evans B, Holt PJ. Successful treatment of pyoderma gangrenosum with topical 0.5% nicotine cream. J Dermatolog Treat 2004; 15:122.
- Wolf R. Nicotine for pyoderma gangrenosum. Arch Dermatol 1998; 134:1071.
- Kanekura T, Kanzaki T. Successful treatment of pyoderma gangrenosum with nicotine chewing gum. J Dermatol 1995; 22:704.
- Nguyen LQ, Weiner J. Treatment of pyoderma gangrenosum with benzoyl peroxide. Cutis 1977; 19:842.
- Vereecken P, Wautrecht JC, De Dobbeleer G, Heenen M. A case of pyoderma gangrenosum stabilized with lymecycline, topical benzoyl peroxide and treated by autograft. Dermatology 1997; 195:50.
- Sanders CJ, Hulsmans RF. Successful treatment of pyoderma gangrenosum with topical 5-aminosalicylic acid. Cutis 1993; 51:262.
- Tsele E, Yu RC, Chu AC. Pyoderma gangrenosum--response to topical nitrogen mustard. Clin Exp Dermatol 1992; 17:437.
- Braun-Falco M, Stock K, Ring J, Hein R. Topical platelet-derived growth factor accelerates healing of myelodysplastic syndrome-associated pyoderma gangrenosum. Br J Dermatol 2002; 147:829.
- Freedman BM, Oplinger EH. Use of becaplermin in progressive limb-threatening pyoderma gangrenosum. Adv Skin Wound Care 2002; 15:180.
- O'Connor C, Gallagher C, Hollywood A, et al. Anakinra for recalcitrant pyoderma gangrenosum. Clin Exp Dermatol 2021; 46:1558.
- Kolios AG, Maul JT, Meier B, et al. Canakinumab in adults with steroid-refractory pyoderma gangrenosum. Br J Dermatol 2015; 173:1216.
- Guénin SH, Khattri S, Lebwohl MG. Spesolimab use in treatment of pyoderma gangrenosum. JAAD Case Rep 2023; 34:18.
- Lee WS, Choi YJ, Yoo WH. Use of tocilizumab in a patient with pyoderma gangrenosum and rheumatoid arthritis. J Eur Acad Dermatol Venereol 2017; 31:e75.
- Scheinberg M, Machado LA, M Castro LG, et al. Successful treatment of ulcerated pyoderma gangrenosum with baricitinib, a novel JAK inhibitor. J Transl Autoimmun 2021; 4:100099.
- Nasifoglu S, Heinrich B, Welzel J. Successful therapy for pyoderma gangrenosum with a Janus kinase 2 inhibitor. Br J Dermatol 2018; 179:504.
- Kochar B, Herfarth N, Mamie C, et al. Tofacitinib for the Treatment of Pyoderma Gangrenosum. Clin Gastroenterol Hepatol 2019; 17:991.
- Orfaly VE, Kovalenko I, Tolkachjov SN, et al. Tofacitinib for the treatment of refractory pyoderma gangrenosum. Clin Exp Dermatol 2021; 46:1082.
- Sedano R, Jairath V. Tofacitinib for the Treatment of Three Immune-mediated Conditions in One Patient: Ulcerative Colitis, Pyoderma Gangrenosum, and Alopecia Areata. Inflamm Bowel Dis 2021; 27:e65.
- Salmón Olavarría P, Rubio Iturria S, Nantes Castillejo Ó. Tofacitinib, a useful option for the treatment of pyoderma gangrenosum in an ulcerative colitis patient. Rev Esp Enferm Dig 2021; 113:733.
- Mensing H. Clofazimine in dermatitis ulcerosa (pyoderma gangrenosum). Open clinical trial. Dermatologica 1988; 177:232.
- Paolini O, Hébuterne X, Flory P, et al. Treatment of pyoderma gangrenosum with colchicine. Lancet 1995; 345:1057.
- Kontochristopoulos GJ, Stavropoulos PG, Gregoriou S, Zakopoulou N. Treatment of Pyoderma gangrenosum with low-dose colchicine. Dermatology 2004; 209:233.
- Smith JB, Shenefelt PD, Soto O, Valeriano J. Pyoderma gangrenosum in a patient with cryoglobulinemia and hepatitis C successfully treated with interferon alfa. J Am Acad Dermatol 1996; 34:901.
- Jones RR, Kobza Black A, Donaghy M, et al. Defective monocyte function in pyoderma gangrenosum with IgG kappa paraproteinaemia. Clin Exp Immunol 1983; 52:685.
- East-Innis A, Desnoes R, Thame K, et al. Pyoderma gangrenosum associated with osteomyelitis in a paediatric patient: a case report. West Indian Med J 2005; 54:207.
- Sheldon DG, Sawchuk LL, Kozarek RA, Thirlby RC. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg 2000; 135:564.
- Richardson JB, Callen JP. Pyoderma gangrenosum treated successfully with potassium iodide. J Am Acad Dermatol 1993; 28:1005.
- Jolles S, Niclasse S, Benson E. Combination oral and topical tacrolimus in therapy-resistant pyoderma gangrenosum. Br J Dermatol 1999; 140:564.
- Kim TH, Oh SY, Myung SC. Pyoderma gangranosum of the penis. J Korean Med Sci 2009; 24:1200.
- Federman GL, Federman DG. Recalcitrant pyoderma gangrenosum treated with thalidomide. Mayo Clin Proc 2000; 75:842.
- Seishima M, Mizutani Y, Shibuya Y, et al. Efficacy of granulocyte and monocyte adsorption apheresis for three cases of refractory pyoderma gangrenosum. Ther Apher Dial 2007; 11:177.
- Fujimoto E, Fujimoto N, Kuroda K, Tajima S. Leukocytapheresis treatment for pyoderma gangrenosum. Br J Dermatol 2004; 151:1090.
- Kanekura T, Maruyama I, Kanzaki T. Granulocyte and monocyte adsorption apheresis for pyoderma gangrenosum. J Am Acad Dermatol 2002; 47:320.
- Okuma K, Mitsuishi K, Hasegawa T, et al. A case report of steroid and immunosuppressant-resistant pyoderma gangrenosum successfully treated by granulocytapheresis. Ther Apher Dial 2007; 11:387.
- Watanabe Y, Yamada H. Leukocyte adsorption apheresis for the treatment of pyoderma gangrenosum. J Dermatol 2008; 35:792.
- Saracino A, Kelly R, Liew D, Chong A. Pyoderma gangrenosum requiring inpatient management: a report of 26 cases with follow up. Australas J Dermatol 2011; 52:218.
- Powell FC, Schroeter AL, Su WP, Perry HO. Pyoderma gangrenosum: a review of 86 patients. Q J Med 1985; 55:173.
- Abdelrazeq AS, Lund JN, Leveson SH. Pouchitis-associated pyoderma gangrenosum following restorative proctocolectomy for ulcerative colitis. Eur J Gastroenterol Hepatol 2004; 16:1057.
- Pishori T, Qureshi AH. Post-colectomy peristomal pyoderma gangrenosum. J Coll Physicians Surg Pak 2005; 15:121.
- Yanaru-Fujisawa R, Matsumoto T, Nakamura S, et al. Granulocyte apheresis for pouchitis with arthritis and pyoderma gangrenosum after restorative proctocolectomy for ulcerative colitis: a case report. Inflamm Bowel Dis 2005; 11:780.
- Holmlund DE, Wählby L. Pyoderma gangrenosum after colectomy for inflammatory bowel disease. Case report. Acta Chir Scand 1987; 153:73.
- Cox NH, Peebles-Brown DA, MacKie RM. Pyoderma gangrenosum occurring 10 years after proctocolectomy for ulcerative colitis. Br J Hosp Med 1986; 36:363.
- Ruocco E, Sangiuliano S, Gravina AG, et al. Pyoderma gangrenosum: an updated review. J Eur Acad Dermatol Venereol 2009; 23:1008.
- Lee S, Lee JY, Ju HJ, et al. Association of All-Cause and Cause-Specific Mortality Risks With Pyoderma Gangrenosum. JAMA Dermatol 2023; 159:151.
- Kaffenberger BH, Hinton A, Krishna SG. The impact of underlying disease state on outcomes in patients with pyoderma gangrenosum: A national survey. J Am Acad Dermatol 2018; 79:659.













