Un hombre de 25 años con antecedentes de leucemia linfoblástica aguda de células B fue evaluado en el departamento de emergencias de este hospital debido a cambios en la visión.
Diez meses antes de esta presentación, el paciente fue
evaluado por fatiga y disnea de esfuerzo. Las pruebas de laboratorio revelaron
un nivel de hemoglobina de 3,5 g por decilitro (rango de referencia, 13,5 a
17,5) y un recuento de glóbulos blancos de 23 450 por microlitro (rango de referencia,
4000 a 11 000), con 81 % de linfocitos y 4 % de blastos. Se realizó una biopsia
de médula ósea.
El examen de la muestra de biopsia de médula ósea (
Figura 1 ) reveló una médula marcadamente hipercelular con una pérdida de
elementos normales. Había láminas de blastos, que incluían células más grandes
con núcleos de ovalados a irregulares, cromatina fina, nucléolos definidos y
citoplasma escaso, así como blastos de aspecto linfocítico más maduros con
cromatina más condensada. El análisis citogenético reveló un cariotipo anormal:
46,XY,t(9;22)(q34;q11.2)[15]/46,idem,t(2;12)(p13;p13),?del(10)(
p13)[3]/46,idem,del(6)(p21),add(7)(p13)[2]. En la secuenciación de próxima
generación, el análisis SNaPshot identificó una mutación RUNX1 y el análisis de
fusión génica fue positivo para BCR-ABL1 e13a2. Se hizo diagnóstico de leucemia linfoblástica aguda de
células B con BCR-ABL1.

Figura 1. Muestra inicial de biopsia de médula ósea.
Se obtuvo una muestra inicial de biopsia de médula
ósea 10 meses antes de esta presentación. La tinción con hematoxilina y eosina
de la muestra (Panel A) muestra una médula
marcadamente hipercelular. A mayor aumento (Panel B), hay blastos más
grandes con cromatina fina (mitad superior) y blastos más pequeños con
cromatina más condensada (mitad inferior).
Se administró quimioterapia de inducción con
vincristina, doxorrubicina, metotrexato, citarabina y prednisona, junto con
dasatinib y citarabina intratecal, metotrexato e hidrocortisona. Un mes
después, el examen de una muestra de biopsia de médula ósea reveló 7% de
blastos. Se inició tratamiento con blinatumomab y se continuó con dasatinib.
Siete meses antes de esta presentación, el examen de
una muestra de biopsia de médula ósea reveló una respuesta hematológica
completa. BCR-ABL1 fue indetectable mediante la prueba de reacción en cadena de
la polimerasa con transcriptasa inversa.
Cinco meses antes de esta presentación, el paciente se
sometió a un trasplante alogénico de células hematopoyéticas (HCT) de un
donante no emparentado compatible después de un acondicionamiento mieloablativo
con ciclofosfamida e irradiación corporal total. Las complicaciones
postrasplante tempranas incluyeron mucositis severa y neutropenia febril.
Dos meses antes de esta presentación, el examen de una
muestra de biopsia de médula ósea reveló una respuesta hematológica completa,
sin reordenamiento de BCR-ABL1 y quimerismo de donante completo. Dasatinib se
continuó como terapia de mantenimiento. Durante los siguientes 2 meses, el
paciente tuvo un empeoramiento de la anemia y se suspendió el dasatinib.
Dos días antes de esta presentación, el paciente notó
“grandes manchas negras” que parecían sombras en la periferia de los campos
visuales de ambos ojos. También hubo episodios intermitentes de visión borrosa en
ambos ojos que se resolvieron después de 10 minutos. Dos días después, el
paciente buscó una evaluación en la clínica de oncología de este hospital y fue
trasladado al servicio de urgencias para una evaluación adicional.
En el departamento de emergencias, el paciente informó
síntomas continuos de la visión, pero no dolor en los ojos, luces intermitentes
ni moscas volantes. También informó 3 semanas de sudores nocturnos, áreas de
hinchazón en el cuello y la ingle y una erupción cutánea pruriginosa en la
parte superior del pecho, la parte superior de la espalda y ambos brazos. Tenía
antecedentes de migrañas diarias crónicas, que con frecuencia se habían
asociado con dolor ocular y fotofobia, pero no se habían asociado con cambios
en la visión. Otros antecedentes médicos incluían depresión y obesidad. Los
medicamentos incluyeron tacrolimus, trimetoprim-sulfametoxazol, famciclovir,
omeprazol y escitalopram, así como suplementos de vitamina D y magnesio. No se
conocen alergias a medicamentos. El paciente vivía en un pueblo costero de
Nueva Inglaterra con su madre. Anteriormente había trabajado en la industria de
servicios de alimentos. Nunca había fumado tabaco ni usado drogas ilícitas;
bebía tres latas de cerveza a la semana. Su historial familiar incluía cáncer
de mama y dolores de cabeza en su abuela materna, cáncer de próstata en su
abuelo paterno e hipertensión en su padre.
En el examen, la temperatura era de 36,9°C, la presión
arterial de 136/88 mm Hg, el pulso de 104 latidos por minuto, la frecuencia
respiratoria de 16 por minuto y la saturación de oxígeno del 100% mientras el
paciente respiraba aire ambiente. La agudeza visual fue de 20/25 en ambos ojos
con corrección y los campos visuales estaban completos en la prueba de
confrontación. Las pupilas eran simétricas y reactivas a la luz. No había
proptosis. El examen con lámpara de hendidura reveló pocas células en la cámara
anterior y el examen de fondo de ojo fue normal. Los resultados de las pruebas
de función, fuerza, sensación, reflejos, coordinación y marcha de los nervios
craneales fueron normales. Había adenopatías palpables en las regiones
cervical, clavicular, axilar e inguinal. Pápulas discretas estaban presentes en
ambos brazos, y xerosis difusa y una erupción eccematosa estaban presentes en
la parte superior de la espalda y el tórax.
Las pruebas de laboratorio revelaron un recuento de plaquetas de 47 000 por microlitro (rango de referencia, 150 000 a 400 000). Los análisis de sangre para el ADN del citomegalovirus, el virus de Epstein-Barr (EBV), el virus de la varicela-zoster y el virus del herpes humano 8 (HHV-8) fueron negativos. Las pruebas de detección del virus de la inmunodeficiencia humana, la sífilis y la enfermedad de Lyme también fueron negativas. El examen de un frotis de sangre periférica confirmó la presencia de anemia y trombocitopenia; no había esquistocitos. En la Tabla 1 se muestran resultados de pruebas de laboratorio adicionales . Se obtuvieron estudios de imagen.

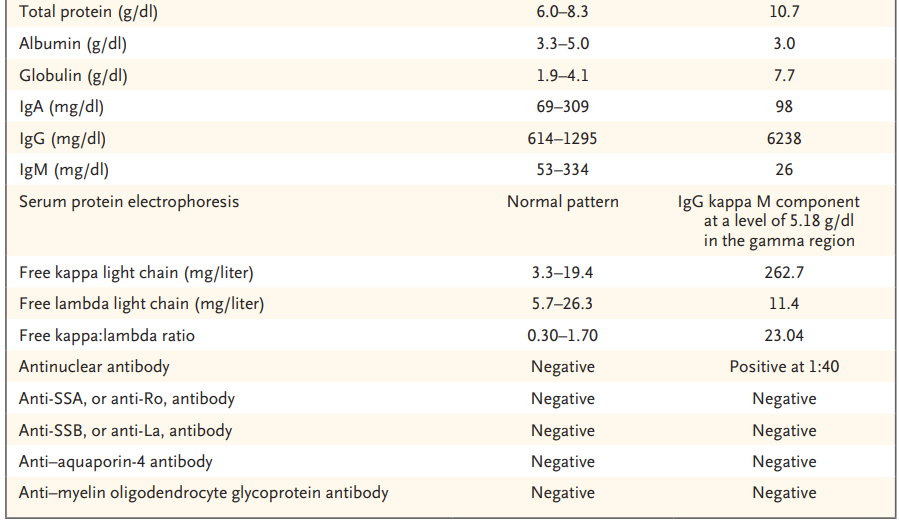
Tabla 1. Datos de laboratorio.
Se realizó una resonancia magnética nuclear (RMN) de
la cabeza. Las imágenes de recuperación de inversión atenuadas por líquido
potenciadas en T2 ( Figura 2A y 2B ) mostraron múltiples lesiones hiperintensas
nuevas, que medían hasta 11 mm en su dimensión mayor, dentro de las regiones
supratentorial e infratentorial del cerebro. Las imágenes obtenidas tras la
administración de material de contraste mostraron un leve realce de las
lesiones. Estos focos parecían estar centrados dentro de la sustancia blanca
yuxtacortical y periventricular y podrían reflejar áreas de desmielinización,
enfermedad de injerto contra huésped (EICH) que afecta al sistema nervioso
central (SNC), leucemia linfoblástica aguda con afectación del SNC o un proceso
infeccioso. No había realce anormal de los nervios ópticos ni de las
estructuras orbitarias.
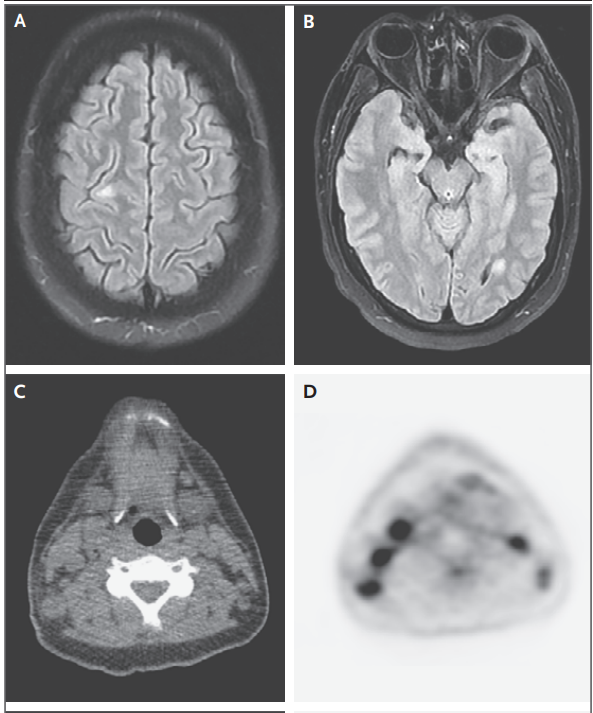

Figura 2. Estudios de imagen obtenidos al ingreso.
Se realizó resonancia magnética al ingreso en este
hospital. Una secuencia ponderado en T2 FLAIR (Paneles A y B) muestran lesiones
hiperintensas en la circunvolución frontal derecha y el lóbulo occipital
izquierdo. Una tomografía por emisión de positrones con 18F‑fluorodesoxi‑
glucosa (PET- FDG), y una TC también fue realizada. Las imágenes PET se renderizaron en tejido
blando (Paneles C, E y G) y escala de grises invertida (paneles D, F y H).
Imágenes axiales de PET‑CT en el nivel cervical (Paneles C y D) muestran
linfadenopatía cervical bilateral ávida de FDG. Las imágenes axiales de PET‑CT
a nivel axilar (paneles E y F) muestran linfadenopatía axilar ávida de FDG que
es mayor en el lado derecho que en el lado izquierdo. Imágenes axiales de PET‑CT
a nivel pélvico (Paneles G y H), muestran linfadenopatía inguinal bilateral ávida de FDG
También se realizó tomografía por emisión de
positrones (PET) y tomografía computarizada (TC) con 18 F-fluorodesoxiglucosa
(FDG). Las exploraciones PET-CT ( Figura 2C a 2H ) mostraron captación de FDG
de moderada a intensa asociada con linfadenopatía generalizada, incluidos los
ganglios linfáticos cervicales, axilares, ilíacos externos e inguinales
bilaterales. También había captación anormal en el bazo.
El paciente ingresó en este hospital y se realizó una
punción lumbar. El análisis del líquido cefalorraquídeo reveló un nivel de
proteína total de 147 mg por decilitro (rango de referencia, 5 a 55) y un nivel
de glucosa de 49 mg por decilitro ( rango de referencia, 50 a 75 mg por decilitro).
El recuento de glóbulos blancos fue de 21 por microlitro (rango de referencia,
0 a 5), con 61 % de linfocitos, 27 % de neutrófilos y 3 % de eosinófilos; el
recuento de glóbulos rojos fue de 16.000 por microlitro (rango de referencia, 0
a 5), con presencia de xantocromía. La tinción de Gram del líquido
cefalorraquídeo mostró una cantidad moderada de células mononucleares y ningún
organismo. Los estudios citológicos fueron notables por el aumento de
linfocitos y células plasmáticas. Se realizó una prueba diagnóstica.
DIAGNÓSTICO DIFERENCIAL
Este hombre de 25 años con antecedentes de leucemia
linfoblástica aguda de células B se presentó 5 meses después de un TCH
alogénico con cambios en la visión, linfadenopatía, empeoramiento de la anemia,
trombocitopenia y detección de proteína monoclonal en la electroforesis de
proteína sérica. La resonancia magnética de la cabeza mostró múltiples lesiones
en la sustancia blanca periventricular. Al desarrollar un diagnóstico
diferencial, consideraré las enfermedades que probablemente ocurran en este paciente,
dada su historia de leucemia linfoblástica aguda y TCH alogénico subsiguiente.
RECAÍDA DE LEUCEMIA AGUDA
Aunque los resultados del trasplante en pacientes
adultos con leucemia linfoblástica aguda han mejorado sustancialmente en las
últimas décadas, 1 la principal causa de TCH fallido sigue siendo la recaída de
la enfermedad (incidencia acumulada, 30 a 40%). 2La recaída de la enfermedad
puede ocurrir en cualquier momento, pero es más probable que ocurra dentro del
primer año después del TCH. Este paciente se presentó 5 meses después del
trasplante con empeoramiento de la anemia, trombocitopenia, linfadenopatía y
lesiones del SNC. Estas características podrían atribuirse a la recaída de la
leucemia linfoblástica aguda con compromiso de la médula ósea y manifestaciones
extramedulares, pero esto es poco común. En general, la recaída de la
enfermedad con compromiso del SNC ocurre con poca frecuencia después de un TCH
alogénico (incidencia acumulada, 2 a 3 %) y es más probable que ocurra en
pacientes con compromiso previo del SNC, 3que no había estado presente en este
paciente. Sin embargo, la recaída de la enfermedad sigue siendo una
posibilidad, y se realizaría una biopsia de médula ósea para evaluar la
evidencia de recaída si no hubiera una explicación alternativa para su
presentación.
INFECCIÓN
Una infección oportunista que afecta al SNC es una
consideración importante en un paciente que presenta cambios agudos en la
visión dentro del primer año después de un TCH alogénico. Los receptores de HCT
alogénico permanecen inmunocomprometidos después del trasplante hasta la
recuperación del sistema inmunitario derivado del donante. El grado y la
duración de la inmunosupresión varían y pueden verse influidos por
características como la fuente del donante y el uso de un régimen
inmunosupresor, así como el desarrollo y tratamiento de la GVHD crónica. 4
Cinco meses después del trasplante, este paciente
seguía en tratamiento con tacrolimus y presentaba riesgo de complicaciones
infecciosas, a pesar de la profilaxis con trimetoprima-sulfametoxazol y
famciclovir. Varias infecciones pueden causar lesiones masivas en el cerebro,
incluidas las infecciones por hongos, bacterias y protozoos. 5 Este paciente
estuvo en riesgo de infección por hongos o moho durante los períodos de
neutropenia prolongada que ocurrieron mientras recibía tratamiento para la
leucemia antes del trasplante. La inmunosupresión continua podría permitir el
desarrollo de una enfermedad clínica fulminante, a pesar de la recuperación de
los neutrófilos. Además, la falta de reconstitución inmune podría contribuir a
la reactivación de una infección previa por Toxoplasma gondii. Sin embargo, es
poco probable que estos patógenos expliquen la linfadenopatía generalizada
observada en la PET-CT, así como la anemia, la trombocitopenia y la proteína
monoclonal.
MEDICAMENTOS
¿Podría alguno de los medicamentos de este paciente
explicar su presentación actual? Tomaba tacrolimus, que se ha asociado con
microangiopatía trombótica 6 y síndrome de encefalopatía posterior reversible.
7Se debe considerar la microangiopatía trombótica asociada al trasplante porque
el paciente tenía una nueva trombocitopenia; este trastorno a menudo se pasa
por alto, ya que sus características clínicas se asemejan a las de otros
trastornos que ocurren después del trasplante. Sin embargo, la ausencia de
esquistocitos en el frotis de sangre periférica hace que la microangiopatía
trombótica asociada con el trasplante sea poco probable. Además, este
diagnóstico no explicaría la linfadenopatía o las lesiones del SNC. Los
pacientes con síndrome de encefalopatía posterior reversible pueden presentar
dolor de cabeza, convulsiones, alteración del estado mental y pérdida de la
visión, junto con cambios asociados en las imágenes. Sin embargo, los hallazgos
de la resonancia magnética en este paciente no fueron consistentes con este
diagnóstico. Además, las complicaciones del tacrolimus no explicarían la
linfadenopatía generalizada.
ENFERMEDAD DE INJERTO CONTRA HUÉSPED
La GVHD es una de las complicaciones inmunológicas más
comunes que ocurren después de un TCH alogénico. La GVHD aguda generalmente
ocurre temprano después del trasplante y afecta principalmente la piel, el
hígado y el tracto gastrointestinal. 8 La GVHD crónica clásicamente ocurre más
adelante en el curso del trasplante y puede manifestarse como cambios
inflamatorios o fibróticos en una amplia gama de órganos diana. 9 A los 5 meses
después del trasplante, sería posible que se desarrollara la GVHD aguda tardía,
la GVHD crónica clásica o un subtipo superpuesto con características tanto
agudas como crónicas. La GVHD puede causar trombocitopenia 10pero no se asocia
comúnmente con linfadenopatía o enfermedad del SNC. Además, este paciente tenía
un sarpullido pero no tenía otros síntomas típicos de la GVHD, como ojos o boca
secos, diarrea, dificultad para tragar, debilidad muscular o tensión en las
articulaciones.
CÁNCER SECUNDARIO
Los hallazgos de imagen en este paciente, incluidas la
linfadenopatía y las lesiones del SNC, podrían representar un cáncer secundario,
como una enfermedad mieloide sólida o extramedular relacionada con el
tratamiento. Estos cánceres pueden ocurrir después del trasplante,
especialmente cuando el trasplante se realiza con irradiación corporal total.
11 Sin embargo, son raros, particularmente dentro del primer año.
TRASTORNO LINFOPROLIFERATIVO POSTRASPLANTE
El trastorno linfoproliferativo postrasplante (PTLD)
es una complicación rara tras un TCH alogénico (127 casos en 26.901 receptores
de trasplante) 12 ; es más probable que ocurra después de un trasplante de
órganos sólidos. 13 PTLD representa un espectro de estados linfoproliferativos
clínicos y morfológicos, que van desde la hiperplasia policlonal reactiva
benigna hasta el linfoma. La mayoría de los casos de PTLD después de un TCH están
asociados con una infección por EBV de origen donante y ocurren dentro del
primer año después del trasplante. 14En el contexto de una inmunidad mediada
por células T deteriorada, las células infectadas por EBV previamente latentes
pueden proliferar y contribuir a la patogenia de PTLD. Los pacientes con PTLD a
menudo presentan fiebre, linfadenopatía y manifestaciones específicas de
órganos. La enfermedad del SNC es rara. Los factores de riesgo incluyen el uso
de depleción de células T ex vivo, la presencia de anticuerpos anti-células T,
el uso de sangre del cordón umbilical como fuente de injerto y la presencia de
incompatibilidad HLA, ninguno de los cuales fue factor en este paciente.
Cuando los pacientes con PTLD tienen una prueba
negativa para EBV, se considera la siguiente causa viral más común, HHV-8.
15Este paciente tuvo análisis de sangre negativos tanto para EBV como para ADN
de HHV-8. Sin embargo, su linfadenopatía difusa fue más consistente con un
diagnóstico de PTLD, por lo que se consideraron otras causas de PTLD. Los
hallazgos de laboratorio notables en la presentación incluyeron niveles
sanguíneos elevados de globulina e IgG; estos hallazgos impulsaron la
electroforesis de proteínas séricas, que mostró proteína monoclonal (componente
IgG kappa M a un nivel de 5,18 g por decilitro en la región gamma). Este
hallazgo fue bastante inesperado y, en el contexto de los hallazgos de PET-CT,
sugería PTLD relacionado con mieloma de células plasmáticas o linfoma no
Hodgkin con proteína monoclonal asociada. Dado el posible diagnóstico de PTLD,
optamos por proceder con la biopsia de un ganglio linfático en la región
inguinal, que era el área de más fácil acceso con linfadenopatía.
DIAGNÓSTICO PRESUNTIVO
TRASTORNO LINFOPROLIFERATIVO POSTRASPLANTE.
DISCUSIÓN PATOLÓGICA
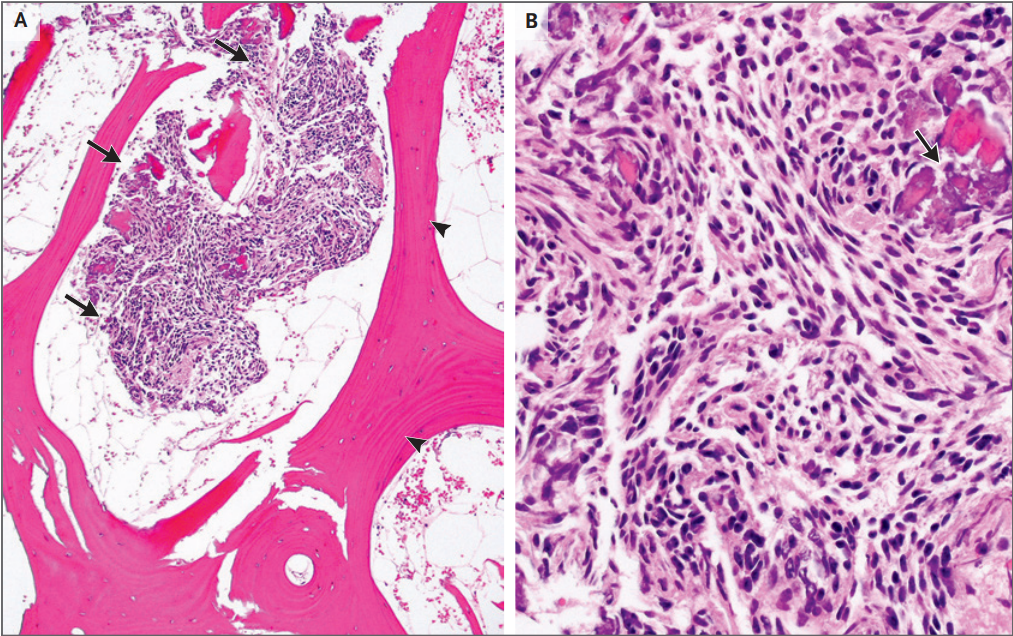
Se obtuvo una muestra de biopsia de un ganglio
linfático inguinal derecho agrandado ( Figura 3 ). La mayor parte de la
arquitectura normal fue borrada por una proliferación difusa de células
plasmáticas y de inmunoblastos ocasionales. La tinción inmunohistoquímica
mostró unos pocos folículos linfoides residuales pequeños, que fueron
resaltados por el marcador de células B CD20. La mayoría de las células eran
células plasmáticas CD138+. En la tinción de cadenas ligeras de inmunoglobulina
kappa y lambda, las células plasmáticas expresaron kappa monotípica. En la
tinción de cadenas pesadas, las células plasmáticas positivas para kappa
coexpresaron la cadena pesada gamma (IgG monotípica kappa). El índice de
proliferación Ki-67 fue de aproximadamente 30%. La hibridación in situ para EBV
fue negativa.
Estos hallazgos son compatibles con un plasmocitoma
ganglionar. Dado el estado posterior al trasplante del paciente y la naturaleza
multifocal de su enfermedad, los hallazgos se clasifican mejor como mieloma de
células plasmáticas PTLD. La hibridación in situ con fluorescencia no reveló
ningún reordenamiento de BCR-ABL1 . Las huellas dactilares de ADN con el uso de
pruebas de quimerismo, realizadas sobre una base de investigación, sugirieron
que el PTLD era de origen receptor. Sin embargo, en ausencia de reordenamiento
de BCR-ABL1 , era poco probable que el PTLD estuviera relacionado clonalmente
con la leucemia linfoblástica aguda de células B previamente diagnosticada.


Figura 3. Muestra de biopsia de un ganglio linfático
inguinal derecho.
Se obtuvo una muestra de biopsia de un ganglio
linfático inguinal derecho durante la ad‑ misión a este hospital. Tinción con
hematoxilina y eosina de la muestra (Panel A) muestra el borramiento arquitectónico.
A mayor aumento (Panel B), predominan las células plasmáticas y pocos
inmunoblastos con cromatina vesicular y nucléolos centrales prominentes.
Tinción inmunohistoquímica para CD20 (Panel C) muestra un folículo linfoide
residual (flecha). La tinción para CD138 (Panel D) resalta las lámminas de
células plasmáticas. Sobre la tinción para cadenas ligeras kappa de
inmunoglobulina y lambda (Paneles E y F,
respectivamente), las células plasmáticas expresan kappa monotípica (Panel E) y
solo células raras expresan lambda (Panel F). En la tinción de cadenas pesadas
(Panel G), las células plasmáticas kappa‑positivas, coexpresan cadenas pesadas
gamma (IgG kappa monotípica). La tinción para Ki‑67 (Panel H) resalta el 30 %
de las células.
DIAGNÓSTICO PATOLÓGICO
TRASTORNO LINFOPROLIFERATIVO POSTRASPLANTE DE MIELOMA
DE CÉLULAS PLASMÁTICAS.
DISCUSIÓN DEL MANEJO
Dada la relativa rareza y heterogeneidad de PTLD,
faltan datos de ensayos aleatorizados para guiar el enfoque del tratamiento
para PTLD después de HCT, especialmente para PTLD de mieloma de células
plasmáticas. En el tratamiento de este paciente, que tenía PTLD después de un
TCH alogénico que era EBV negativo, monomórfico, derivado del receptor y no
relacionado con el linfoma difuso de células B grandes, consideré tres
elementos esenciales: disminuir el curso de la inmunosupresión sistémica, proporcionar
tratamiento dirigido a la enfermedad y administración de una infusión de
linfocitos de donante.
La supresión del sistema inmunitario del huésped es
uno de los principales factores determinantes en la fisiopatología de la PTLD
y, a menudo, se recomienda reducir la dosis de agentes inmunosupresores como
paso inicial del tratamiento. 14 En pacientes que se han sometido a un TCH
alogénico, no siempre es posible reducir rápidamente la inmunosupresión debido
al deseo competitivo de prevenir la aparición de EICH aguda y crónica. La
probabilidad de que el PTLD se resuelva después de la reducción gradual de la
inmunosupresión sola varía ampliamente y la mayoría de los pacientes reciben
terapia adicional.
Al determinar el tratamiento apropiado dirigido a la
enfermedad para este paciente, se debe considerar el proceso de células
plasmáticas. Las características de su presentación eran similares a las del
mieloma múltiple extramedular, con un gran número de lesiones ganglionares y
extraganglionares y sin afectación sustancial de la médula ósea. Aunque la
radioterapia local es un pilar del tratamiento del plasmacitoma solitario, 16la
radioterapia no tenía ningún papel en este paciente, dado que tenía múltiples
áreas de afectación, que no tenía una lesión específica que estuviera causando
síntomas localizados severos y que requiriera una intervención urgente, y que
no tenía una lesión a la que la podrían atribuirse cambios en la visión. Los
regímenes múltiples que incluyen una combinación de tres o cuatro fármacos
están disponibles para la terapia sistémica, debido a los avances en la
terapéutica del mieloma en los últimos años. 17
Inicialmente, elegimos tratar a este paciente con
daratumumab (un anticuerpo monoclonal anti-CD38) como agente único, 18 una
terapia asociada con un potencial de tratamiento dirigido, efectos tóxicos
limitados e informes de seguridad cuando se usa después de un TCH alogénico. 19
El nivel de proteína monoclonal sérica disminuyó con un ciclo de terapia, pero
desafortunadamente, la trombocitopenia empeoró y una exploración PET-CT
repetida mostró un aumento en el tamaño de los ganglios linfáticos
involucrados, aunque con una menor avidez de FDG. Posteriormente tratamos al
paciente con una combinación de ciclofosfamida, bortezomib y dexametasona. 20
Elegimos evitar los regímenes que incluían inmunomoduladores (p. ej.,
lenalidomida), dada su asociación con la GVHD cuando se usan después de un TCH
alogénico. 21,22
Después de que el paciente recibió un ciclo de
daratumumab y completó seis ciclos de una combinación de ciclofosfamida,
bortezomib y dexametasona, los cambios en la visión disminuyeron y hubo una
respuesta radiológica casi completa evaluada mediante resonancia magnética y
PET-TC en serie ( Figura 4 ). Aunque los cambios en la visión no podían
explicarse por la ubicación anatómica de las lesiones del SNC, se resolvieron
con la respuesta de la enfermedad. En consecuencia, el nivel de proteína
monoclonal IgG kappa disminuyó de 5,18 a 0,07 g por decilitro y mejoraron los
recuentos de sangre periférica.

Figura 4. Exploraciones PET obtenidas antes y después
del tratamiento.
Una tomografía por emisión de positrones (PET)
obtenida al ingreso (Panel A) muestra niveles múltiples de linfadenopatías
ávidas de FDG, más pronunciada en las regiones de axila e inguinal. Una
tomografía PET obtenida después del tratamiento (Panel B) muestra resolución de
la linfadenopatía ávida de FDG y sin nuevos sitios de enfermedad.
Finalmente, existe un papel potencial para la infusión
de linfocitos del donante como una forma adicional de terapia celular en el
tratamiento de la PTLD, principalmente cuando la enfermedad responde a la
terapia y se ha demostrado que se origina en el receptor. Sin embargo, la
infusión de linfocitos del donante se asocia con un riesgo de inducir GVHD,
especialmente si el curso de la inmunosupresión sistémica se ha reducido con
bastante rapidez. 23 Los informes de resultados clínicos con el uso de infusión
de linfocitos de donantes para tratar PTLD después de TCH son limitados, pero
las tasas de respuesta en esta situación clínica pueden ser altas (70%). 24Este
paciente recibió tres infusiones de linfocitos de donantes con dosis escaladas
con el objetivo de aumentar la probabilidad de una respuesta al tratamiento a
largo plazo. Posteriormente se desarrolló GVHD grave del hígado y ha respondido
a la terapia. Permanece en remisión completa de PTLD, con un nivel indetectable
de proteína monoclonal y sin necesidad de terapia dirigida a la enfermedad
durante 6 meses.
DIAGNOSTICO FINAL
TRASTORNO LINFOPROLIFERATIVO POSTRASPLANTE DE MIELOMA
DE CÉLULAS PLASMÁTICAS.
Traducción de
A 25-Year-Old Man with Vision Changes
Zachariah DeFilipp, M.D., Karen Rodriguez, M.D., and
Judith A. Ferry, M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2115854?query=featured_home
References
1. Center for International Blood and
Marrow Transplant Research. The US sum[1]mary
slides — HCT trends and survival
data (https://www . cibmtr . org/ Reference
Center/ SlidesReports/ SummarySlides/ pages/
index . aspx).
2. Kebriaei P, Anasetti C, Zhang M-J, et al.
Intravenous busulfan compared with total
body irradiation pretransplant condition[1]ing
for adults with acute lymphoblastic
leukemia. Biol Blood Marrow Transplant
2018; 24: 726-33.
3. Oshima K, Kanda Y, Yamashita T, et al.
Central nervous system relapse of leuke[1]mia
after allogeneic hematopoietic stem
cell transplantation. Biol Blood Marrow
Transplant 2008; 14: 1100-7.
4. Ogonek J, Kralj Juric M, Ghimire S,
et al. Immune reconstitution after alloge[1]neic
hematopoietic stem cell transplanta[1]tion.
Front Immunol 2016; 7: 507.
5. Castro I, Ruiz J, Tasias M, Montero M,
Salavert M. Central nervous system infec[1]tions
in immunocompromised patients.
Rev Esp Quimioter 2018; 31: Suppl 1: 56-
61.
6. Gavriilaki E, Sakellari I, Anagnosto[1]poulos
A, Brodsky RA. Transplant-asso[1]ciated
thrombotic microangiopathy:
opening Pandora’s box. Bone Marrow
Transplant 2017; 52: 1355-60.
7. Hodnett P, Coyle J, O’Regan K, Maher
MM, Fanning N. PRES (posterior revers[1]ible
encephalopathy syndrome), a rare
complication of tacrolimus therapy. Emerg
Radiol 2009; 16: 493-6.
8. Zeiser R, Blazar BR. Acute graft-ver[1]sus-host
disease — biologic process, pre[1]vention,
and therapy. N Engl J Med 2017;
377: 2167-79.
9. Zeiser R, Blazar BR. Pathophysiology
of chronic graft-versus-host disease and
Figure 4. PET Scans Obtained before and after
Treatment.
A PET scan obtained on admission (Panel A) shows
multilevel FDG‑avid
lymphadenopathy, which is most pronounced in the
axillary and inguinal
regions. A PET scan obtained after treatment (Panel B)
shows resolution
of the FDG‑avid lymphadenopathy and no new sites of
disease.
377:2565-79.
10. Pulanic D, Lozier JN, Pavletic SZ.
Thrombocytopenia and hemostatic disor[1]ders
in chronic graft versus host disease.
Bone Marrow Transplant 2009;44:393-403.
11. Lee CJ, Kim S, Tecca HR, et al. Late
effects after ablative allogeneic stem cell
transplantation for adolescent and young
adult acute myeloid leukemia. Blood Adv
2020;4:983-92.
12. Landgren O, Gilbert ES, Rizzo JD,
et al. Risk factors for lymphoproliferative
disorders after allogeneic hematopoietic
cell transplantation. Blood 2009;113:4992-
5001.
13. Opelz G, Döhler B. Lymphomas after
solid organ transplantation: a collabora[1]tive
transplant study report. Am J Trans[1]plant
2004;4:222-30.
14. Dierickx D, Habermann TM. Post[1]transplantation
lymphoproliferative dis[1]orders
in adults. N Engl J Med 2018;378:
549-62.
15. Riva G, Luppi M, Barozzi P, Forghieri
F, Potenza L. How I treat HHV8/KSHV[1]related
diseases in posttransplant patients.
Blood 2012;120:4150-9.
16. Fotiou D, Dimopoulos MA, Kastritis
E. How we manage patients with plasma[1]cytomas.
Curr Hematol Malig Rep 2018;
13:227-35.
17. Dimopoulos MA, Moreau P, Terpos E,
et al. Multiple myeloma: EHA-ESMO clini[1]cal
practice guidelines for diagnosis, treat[1]ment and follow-up†.
Ann Oncol 2021;32:
309-22.
18. Lokhorst HM, Plesner T, Laubach JP,
et al. Targeting CD38 with daratumumab
monotherapy in multiple myeloma. N Engl
J Med 2015;373:1207-19.
19. Chapuy CI, Kaufman RM, Alyea EP,
Connors JM. Daratumumab for delayed
red-cell engraftment after allogeneic trans[1]plantation.
N Engl J Med 2018;379:1846-
50.
20. Reeder CB, Reece DE, Kukreti V, et al.
Cyclophosphamide, bortezomib and dexa[1]methasone
induction for newly diagnosed
multiple myeloma: high response rates in
a phase II clinical trial. Leukemia 2009;
23:1337-41.
21. Kneppers E, van der Holt B, Kersten
M-J, et al. Lenalidomide maintenance af[1]ter
nonmyeloablative allogeneic stem cell
transplantation in multiple myeloma is not
feasible: results of the HOVON 76 trial.
Blood 2011;118:2413-9.
22. Kröger N, Zabelina T, Klyuchnikov E,
et al. Toxicity-reduced, myeloablative allo[1]graft
followed by lenalidomide mainte[1]nance
as salvage therapy for refractory/
relapsed myeloma patients. Bone Marrow
Transplant 2013;48:403-7.
23. Frey NV, Porter DL. Graft-versus-host
disease after donor leukocyte infusions:
presentation and management. Best Pract
Res Clin Haematol 2008;21:205-22.
24. Doubrovina E, Oflaz-Sozmen B,
Prockop SE, et al. Adoptive immunother[1]apy
with unselected or EBV-specific T cells
for biopsy-proven EBV+ lymphomas after
allogeneic hematopoietic cell transplan[1]tation.
Blood 2012;119:2644-56.