En este ejercicio clínico se presenta un caso que es
discutido por un médico internista al que se le van proporcionando datos de la
historia clínica en forma secuencial, y este analiza el cuadro a la luz de los
nuevos elementos, de una manera análoga al proceso diagnóstico en la práctica
real de la medicina
Una mujer de 32 años acudió al servicio de urgencias
con un historial de empeoramiento de su dolor abdominal de 6 meses de
evolución. Calificó el dolor como 10 en una escala de 10 puntos de gravedad; el
dolor se localizó en el epigastrio y se caracterizó por ser de naturaleza
intermitente y tipo calambre, sin radiación sustancial. No se asoció con comer
o hacer ejercicio, pero se asoció con una pérdida de peso involuntaria de 9 kg
(20 lb). Informó escalofríos, sudores nocturnos, tos no productiva y disnea
intermitente y sibilancias que no respondían al albuterol inhalado. Informó que
no había tenido fiebre, cefaleas, alteraciones de la visión, dolor torácico,
náuseas, emesis, cambios en los hábitos intestinales ni síntomas
genitourinarios. Sus ciclos menstruales eran normales.
PONENTE
Los trastornos gastrointestinales localizados (p. ej.,
gastritis, úlcera péptica y pancreatitis) podrían causar dolor abdominal
crónico, pero no explicarían los síntomas sistémicos de esta paciente.
Escalofríos, sudores y pérdida de peso sugieren una enfermedad sistémica
infecciosa, inflamatoria o neoplásica. Una infección indolente, como
tuberculosis o histoplasmosis, también podría explicar sus síntomas pulmonares.
Un cáncer, como un linfoma, podría causar una constelación similar de síntomas
como resultado de la linfadenopatía pulmonar y abdominal. Los síntomas
adicionales que sugieran una enfermedad autoinmune (fenómeno de Raynaud,
artritis, úlceras orales y cambios en los ojos o la piel), exposiciones
infecciosas y antecedentes familiares de enfermedad inflamatoria o cáncer
ayudarían a refinar el diagnóstico diferencial.
EVOLUCIÓN
La paciente notó dolor articular intermitente en ambas
manos, pero no informó cambios de color en los dedos ni dolor en otras partes
de sus extremidades. Informó que había tenido cambios en la piel de sus piernas
en el transcurso de 6 meses, con protuberancias no dolorosas presentes en los
sitios de tatuajes recientes. No tenía ojos secos, boca seca, úlceras bucales,
dolor torácico pleurítico, fotosensibilidad ni alopecia. Su historial médico
era notable por reflujo gastroesofágico y estreñimiento. No tenía antecedentes
de cirugía. Sus medicamentos incluían paracetamol dos veces al día para el
dolor abdominal, con un efecto modesto. No tenía alergias conocidas. Se
identificó como negra y vivía en Boston; trabajaba como asistente de atención
al paciente y no viajaba fuera de Massachusetts. Era sexualmente activa con una
pareja masculina y usaba condones constantemente. Había fumado medio paquete de
cigarrillos al día durante 13 años, y rara vez usaba marihuana o vapeaba. No
tenía exposiciones conocidas a partículas inhaladas como sílice o polvos
metálicos, incluido el berilio. Consumía ocho bebidas alcohólicas a la semana.
Informó que no consumía otras drogas. No había antecedentes familiares de
cáncer o enfermedad gastrointestinal; una tía tenía sarcoidosis.
PONENTE
El reflujo y el estreñimiento podrían contribuir al
dolor abdominal, pero no explicarían los sudores nocturnos ni la pérdida de
peso. La ausencia de viajes fuera de Massachusetts reduce la preocupación por
una micosis endémica como la histoplasmosis, pero su ocupación en el cuidado de
la salud aumenta el riesgo de tuberculosis. Aunque su consumo de alcohol excede
modestamente la ingesta "saludable" (<7 bebidas por semana en
mujeres), es poco probable que cause complicaciones relacionadas con el alcohol
a esta edad. Los cambios en la piel relacionados con los tatuajes se pueden observar
en condiciones infecciosas e inflamatorias o granulomatosas, más clásicamente
en la sarcoidosis; los antecedentes familiares aumentan la preocupación por la
sarcoidosis.
EVOLUCIÓN
La temperatura oral de la paciente era de 36,5°C, la
frecuencia cardíaca de 62 latidos por minuto, la presión arterial de 122/66 mm
Hg, la frecuencia respiratoria de 16 por minuto y la saturación de oxígeno del
98% mientras respiraba aire ambiente. Su altura era de 163 cm y su peso de 58
kg, para un índice de masa corporal (el peso en kilogramos dividido por el
cuadrado de la altura en metros) de 21,8. Tenía buen aspecto, sin caquexia. Las
conjuntivas, orofaringe y glándulas parótidas eran normales. Había
linfadenopatía no dolorosa bilateralmente en las cadenas cervical,
supraclavicular, axilar e inguinal. Los exámenes cardíaco y pulmonar fueron
normales. El abdomen estaba blando y no distendido, con leve dolor a la
palpación profunda en el epigastrio sin rebote, defensa, organomegalia u otras
masas. Las extremidades mostraban sensibilidad leve a la palpación en el codo
izquierdo y la rodilla derecha, con un rango completo de movimiento y sin
eritema, hinchazón o derrame; las articulaciones restantes no tenían nada
especial. El examen neurológico fue normal. La piel del paciente estaba
caliente y seca; Había pápulas eritematosas y violáceas dispersas, firmes,
ligeramente erosionadas, no dolorosas, en los brazos y las piernas, con agrupamiento
o cluster en la rodilla derecha y tatuajes bilaterales suprayacentes en las
piernas (Figura 1 ).

Figura 1. Cambios
en la piel.
Los paneles A y B muestran cambios en la piel que
recubren los tatuajes en la parte lateral de la pierna derecha (Panel A) y en
la parte posterior de la pierna izquierda (Panel B). El panel C muestra un
grupo de pápulas en la superficie medial de la rodilla derecha.
PONENTE
Aunque la linfadenopatía difusa puede reflejar
procesos infecciosos o inflamatorios, el linfoma es motivo de especial
preocupación. La combinación de artralgias, linfadenopatías y exantemas podría
explicarse por lupus eritematoso sistémico incluso sin fenómeno de Raynaud,
fotosensibilidad o alopecia. La erupción papular eritematosa puede ser causada
por una infección como la borreliosis o una enfermedad micobacteriana, cánceres
como el linfoma cutáneo de la zona marginal o el linfoma de células T, o una
reacción inflamatoria dérmica. La asociación con la tinta del tatuaje es
característica de la sarcoidosis, pero también puede verse en el contexto de un
pseudolinfoma cutáneo (linfoproliferación benigna en respuesta a antígenos
cutáneos) o infecciones relacionadas con la inoculación local.
EVOLUCIÓN
Los resultados de las pruebas de función hepática y
los niveles séricos de electrolitos, creatinina, calcio, lipasa y hormona
estimulante de la tiroides fueron normales, al igual que el tiempo de
protrombina y el tiempo parcial de tromboplastina. El recuento de glóbulos
blancos fue de 5240 por microlitro, con 36,1 % de neutrófilos (rango de
referencia, 48 a 76), 36,8 % de linfocitos (rango de referencia, 18 a 41), 11,6
% de monocitos (rango de referencia, 4 a 11) y 14,5 % eosinófilos (rango de
referencia, 0 a 5); el nivel de hemoglobina era de 13,6 g por decilitro y el
recuento de plaquetas de 338.000 por microlitro. La velocidad de sedimentación
globular era de 50 mm por hora (rango de referencia, 0 a 20) y el nivel de
proteína C reactiva era normal. El nivel de lactato deshidrogenasa fue de 294 U
por litro (rango de referencia, 135 a 225), el nivel de enzima convertidora de
angiotensina de 146 U por litro (rango de referencia, 16 a 85), y el nivel de
IgG total 2243 mg por decilitro (rango de referencia, 700 a 1600). Un ensayo de
liberación de interferón gamma para tuberculosis fue negativo, al igual que la
prueba de inmunoglobulina para sífilis. Las pruebas serológicas para los virus
de la hepatitis A, B y C, el virus de la inmunodeficiencia humana, los
anticuerpos antinucleares, los anticuerpos anticitoplasma de neutrófilos y los
anticuerpos anti-ADN de doble cadena fueron negativos; los niveles de C3 y C4
eran normales. El análisis de orina fue normal y una prueba de embarazo en
orina fue negativa.
´
PONENTE
Muchas de estas pruebas sugieren una mayor activación
no específica del sistema inmunitario. Una velocidad de sedimentación globular
elevada con un nivel normal de proteína C reactiva se puede observar en el
contexto de infecciones, enfermedades autoinmunes como el lupus eritematoso
sistémico y cánceres con sobreproducción de inmunoglobulinas (p. ej.,
macroglobulinemia de Waldenstrom y mieloma múltiple). La eosinofilia puede
ocurrir con cánceres, incluidos los linfomas; en trastornos inflamatorios, como
la sarcoidosis; o en infecciones (típicamente infecciones parasitarias, pero
también otras, incluidas infecciones fúngicas). Un nivel sérico elevado de
enzima convertidora de angiotensina también es inespecífico e inadecuado para
el diagnóstico de sarcoidosis; puede estar elevado en el contexto de linfoma de
Hodgkin, coccidioidomicosis y beriliosis, entre otras condiciones. El ensayo de
liberación de interferón gamma negativo hace que la tuberculosis sea menos
probable, pero este ensayo puede ser falso negativo en pacientes con
tuberculosis diseminada. La negatividad para anticuerpos antinucleares
básicamente descarta el lupus eritematoso sistémico. La ausencia de proteinuria
o hematuria descarta glomerulonefritis. En general, sigo preocupado por el
linfoma, con sarcoidosis como posible diagnóstico alternativo.
EVOLUCIÓN
La tomografía computarizada (TC) de tórax y abdomen
realizada después de la administración de material de contraste ( Figura 2A a
2D ) mostró adenopatías peritoneales y retroperitoneales difusas torácicas y
voluminosas extensas, así como opacidades pulmonares nodulares subcentimétricas
en el lóbulo inferior que eran compatibles con un posible "signo de la
galaxia". ” (es decir, granulomas pulmonares coalescentes que crean una
apariencia de masa) ( Figura 2A ). La tomografía por emisión de positrones
(PET) de cuerpo entero mostró ganglios linfáticos intensamente hipermetabólicos
en las regiones cervical, retrofaríngea, mediastínica, mamaria interna,
subcarinal, axilar, abdominopélvica, periaórtica, mesentérica, ilíaca e
inguinal. Se observaron lesiones hipermetabólicas adicionales en áreas de
hueso, pulmón y piel ( Figura 2E a 2H ).

Figura 2. Tomografías computarizadas y PET de tórax,
abdomen y pelvis.
Las tomografías computarizadas mostraron nódulos
pulmonares coalescentes en el lóbulo superior derecho (marcados con un círculo
en el Panel A), nódulos pulmonares en el lóbulo inferior en ambos pulmones
(Panel B), linfadenopatía mediastínica e hiliar (Panel C) y linfadenopatía
intraabdominal (Panel D). Las tomografías por emisión de positrones (PET)
mostraron linfadenopatía en los ganglios cervicales hipermetabólicos (Panel E);
ganglios axilares, pulmonares y mediastínicos (Panel F); ganglios
intraabdominales y mesentéricos (Panel G); y ganglios inguinales (Panel H). La
flecha en el Panel H indica el ganglio linfático inguinal del que se realizó la
biopsia.
PONENTE
La linfadenopatía extensa es altamente sugestiva de un
trastorno linfoproliferativo, en particular linfoma, y el hipermetabolismo
que se observa en la PET, respalda aún más este diagnóstico. La linfadenopatía
de un tumor sólido metastásico, como el cáncer de mama o de pulmón, es poco
probable en esta paciente joven sin evidencia de un tumor primario. La
linfadenopatía hipermetabólica también puede estar asociada con procesos infecciosos
o inflamatorios, incluidas infecciones bacterianas o fúngicas diseminadas o
respuestas autoinmunes, como en el lupus eritematoso sistémico. La sarcoidosis
sigue siendo posible, dadas las adenopatías mediastínicas e hiliares
bilaterales simétricas, y también podría explicar la afectación cutánea y ósea;
La sarcoidosis también es una causa bien reconocida de un signo de galaxia,
aunque este hallazgo también puede ocurrir en otras afecciones, como la
tuberculosis.
EVOLUCIÓN
El paciente se sometió a una biopsia por escisión de
un ganglio linfático de la ingle derecha. La citometría de flujo de la muestra
de tejido no mostró evidencia de un trastorno linfoproliferativo. El análisis
patológico mostró borramiento de los ganglios linfáticos por linfadenitis
granulomatosa no necrotizante sin células malignas ( Figura 3 ). La tinción
Gram y ácido-resistente fue negativa, al igual que los cultivos bacterianos y
fúngicos.

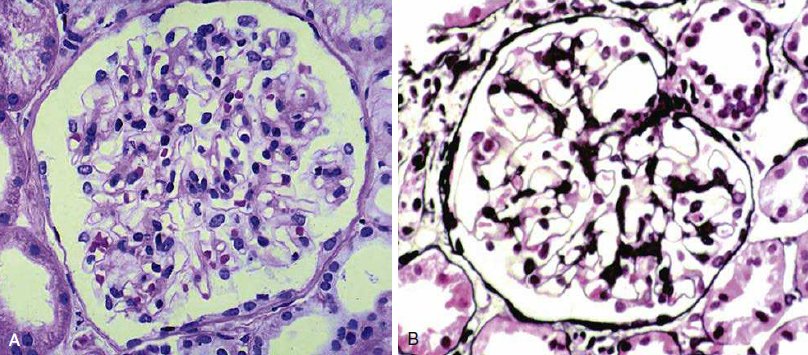
Figura 3. Tejido del ganglio linfático inguinal
derecho con cambios granulomatosos difusos.
La imagen en el Panel B se muestra con un aumento de 5
veces el que se muestra en el Panel A. Las flechas indican granulomas no
necrotizantes individuales.
PONENTE
El borramiento de la arquitectura de los ganglios
linfáticos por granulomas con un núcleo no necrosante es característico, pero
no específico, de la sarcoidosis. Las infecciones por hongos y micobacterias
deben descartarse con cultivos microbiológicos y tinciones. Es posible que se
necesiten múltiples muestras de biopsia para identificar la tuberculosis; sin
embargo, el riesgo de tuberculosis de este paciente es relativamente bajo, y
clásicamente tanto la tuberculosis como la infección fúngica aparecerían
histológicamente como granulomas necrotizantes (caseificantes). Las reacciones
granulomatosas de tipo sarcoide también se pueden observar en asociación con el
linfoma de Hodgkin, los linfomas no Hodgkin, el seminoma en los hombres y una
variedad de carcinomas; la correlación clínica es esencial para excluir esta
posibilidad.En este caso, la combinación de hallazgos histológicos (incluida la
ausencia de cáncer o infección), signos radiográficos y manifestaciones
cutáneas son diagnósticos de sarcoidosis.
EVOLUCIÓN
Se prescribió prednisona a una dosis de 40 mg diarios
y la paciente mejoró rápidamente de sus síntomas abdominales y pulmonares. No
informó palpitaciones, mareos o síncope; un electrocardiograma no mostró
alteraciones de la conducción. Una evaluación oftalmológica no mostró uveítis
ni anomalías retinianas como lesiones de "goteo de cera de vela"
(exudados perivenosos asociados con periflebitis en pacientes con sarcoidosis
ocular aguda). Los cultivos de biopsia fueron negativos para micobacterias
después de 60 días.
La paciente estuvo bien durante 1 año, pero tuvo una
recurrencia de los síntomas después de completar una reducción gradual de
prednisona. Se reinició el tratamiento con prednisona y se pasó al paciente a
metotrexato (un inmunosupresor ahorrador de glucocorticoides).
Comentario
Esta mujer joven se presentó con dolor abdominal,
sudoración y pérdida de peso involuntaria y se encontró que tenía
linfadenopatía difusa, lo que despertó la preocupación por un linfoma. El
diagnóstico diferencial de la linfadenopatía difusa es amplio, pero en última
instancia, el hallazgo de granulomas no necrotizantes en la biopsia de ganglio
linfático por escisión con el descarte de cáncer e infección condujo a un
diagnóstico de sarcoidosis, el inicio de un tratamiento eficaz y la disminución
de los síntomas.
La sarcoidosis se puede diagnosticar con una
presentación clínica apropiada, evidencia de granulomas no necrotizantes en la
biopsia de tejido y descartando otras causas de enfermedad granulomatosa. La
sarcoidosis generalmente involucra los pulmones (en el 95 % de los pacientes en
una serie grande 1 ), la piel (en el 24 %), los ganglios linfáticos (en el 15
%), los ojos (en el 12 %) y el hígado (en el 12 %), pero también puede
involucrar otros órganos. Aunque las manifestaciones clínicas de compromiso
cardíaco son raras, un estudio que involucró autopsias de personas con
sarcoidosis mostró evidencia de granulomas (a menudo microscópicos) en
aproximadamente una cuarta parte de los casos. 2Según un estudio que involucró
una gran base de datos de atención médica de los EE. UU., la incidencia general
de sarcoidosis es de 8 por 100 000 habitantes en los Estados Unidos, pero varía
ampliamente según el sexo, la edad y la raza; Se encontró que las mujeres, las
personas mayores de 45 años y las personas que se identificaron como negras
tenían un riesgo elevado. 3El diagnóstico suele ser un desafío porque muchos
pacientes, como en este caso, tienen una enfermedad que progresa gradualmente
y, a menudo, síntomas inespecíficos. El examen anatomopatológico que muestre
granulomas no necrotizantes y el descarte de cáncer e infección son necesarios
para el diagnóstico de sarcoidosis en la mayoría de los casos; las excepciones
incluyen pacientes con síndrome de Lofgren (una forma de sarcoidosis caracterizada
por fiebre aguda, eritema nodoso, linfadenopatía hiliar bilateral y
periartritis) o síndrome de Heerfordt (también llamado fiebre uveoparotídea,
otra forma de sarcoidosis), en quienes la biopsia puede diferirse. 4
Se debe obtener una muestra de biopsia del sitio más
accesible de presunto compromiso. Histológicamente, los granulomas están bien
formados y tienen necrosis nula o mínima, como se observó en este caso. Los
granulomas están compuestos por macrófagos epitelioides (macrófagos activados
que se asemejan a células epiteliales), células gigantes multinucleadas y
linfocitos T CD4+, a menudo rodeados de infiltrados linfocíticos limitados sin
inflamación neutrofílica pronunciada. Debido a que los granulomas pueden
formarse en respuesta a partículas o patógenos, se necesitan tinciones y
cultivos especiales para descartar otras causas de enfermedad granulomatosa,
incluida la infección por hongos y micobacterias. Se ha propuesto un papel para
los microbios en la instigación de la reacción granulomatosa en la sarcoidosis.
5Las reacciones granulomatosas de tipo sarcoide también pueden ocurrir en
cánceres, incluidos el cáncer de pulmón, el cáncer de mama y los linfomas. 6
Las lesiones cutáneas en los sitios del tatuaje, como
en este paciente, son características de la sarcoidosis; pueden ocurrir en
respuesta a pigmentos específicos 7 y pueden desarrollarse mucho después de
colocarse un tatuaje. Las lesiones también pueden ocurrir en sitios de trauma
("sarcoidosis cicatricial"). Otras manifestaciones dermatológicas
incluyen eritema nodoso (nódulos subcutáneos sensibles debido a paniculitis, a
menudo sobre las espinillas), pápulas faciales cerosas y lupus pernio (placas
violáceas crónicas, a menudo en la nariz y las mejillas). 8
La sarcoidosis pulmonar a menudo aparece
radiográficamente como enfermedad pulmonar intersticial o nódulos pulmonares
(granulomas). Estas anomalías en las imágenes suelen predominar en el lóbulo
superior, a diferencia de las anomalías de este caso. Las anomalías pulmonares
a menudo muestran un engrosamiento del tabique peribronquial e interlobulillar
en un patrón que sigue a los tractos linfáticos. 9 Los nódulos pulmonares
observados en pacientes con sarcoidosis pueden mostrar coalescencia, en un
patrón conocido como signo de galaxia, como se observa en las imágenes de este
paciente, aunque en este paciente había menos lesiones satélite alrededor de la
masa que las que se observan clásicamente (Figura 2A ) . 10
Después del diagnóstico de sarcoidosis, se debe
realizar una evaluación de otros sitios de afectación, incluidos el corazón y
los ojos. La afectación cardíaca es una de las principales causas de muerte,
relacionada con las arritmias. Como se hizo en este caso, los pacientes deben
someterse a un examen electrocardiográfico y evaluar síntomas como
palpitaciones, mareos y síncope. Si existe preocupación acerca de la afectación
cardíaca, se debe buscar la monitorización Holter, la ecocardiografía y,
posiblemente, imágenes adicionales mediante resonancia magnética cardíaca o
PET. Debido a que la afectación ocular puede ser asintomática, todos los
pacientes deben someterse a una evaluación oftalmológica. 11 Directrices
recientes 4También recomienda la detección de calcio sérico para el metabolismo
anormal del calcio causado por la activación de la vitamina D por parte de los
macrófagos, lo que puede provocar lesión renal y aumento de la resorción ósea.
Las imágenes a menudo revelan compromiso asintomático
de otros tejidos, incluidos huesos y músculos, como se observó en este
paciente. La afectación ósea puede afectar el esqueleto axial, las manos y los
pies, así como la artritis de las articulaciones grandes y pequeñas, 12 que
pueden haber causado las artralgias de este paciente. Cuando se toman muestras,
el líquido sinovial muestra un recuento de glóbulos blancos moderadamente
elevado y el tejido sinovial muestra infiltrados de células mononucleares
inespecíficos, aunque no necesariamente granulomas. 13
El tratamiento de la sarcoidosis no es necesario de
forma rutinaria, pero está indicado en personas con síntomas progresivos o
lesión de órganos. Dada la extensión de los síntomas de esta paciente y la
linfadenopatía sospechosa de causar su dolor abdominal, se inició el
tratamiento con terapia con glucocorticoides. Los datos de ensayos clínicos y
las guías de consenso respaldan el uso de primera línea de glucocorticoides,
generalmente prednisona en una dosis de 20 a 40 mg diarios, que generalmente se
reduce después de 3 a 6 meses (o antes si ocurren efectos adversos
importantes). 14 Si el tratamiento con glucocorticoides no puede reducirse con
éxito, se pueden agregar agentes inmunosupresores como metotrexato, inhibidores
del factor de necrosis tumoral u otros medicamentos (micofenolato de mofetilo,
leflunomida o azatioprina).
Este caso destaca el amplio diagnóstico diferencial de
la linfadenopatía difusa, el valor de la toma de muestras de tejido y la
importancia de considerar la sarcoidosis en el diagnóstico diferencial. En este
caso, inicialmente se sospechó fuertemente de linfoma, pero los resultados del
examen anatomopatológico descartaron este diagnóstico y apuntaron a la
sarcoidosis como la causa de las manifestaciones multisistémicas de este
paciente.
Traducido de:
A Bumpy Road to Diagnosis
Jacob J. Cedarbaum, M.D., M.S.Ed., Deepak A. Rao,
M.D., Ph.D., Hiroto Hatabu, M.D., Ph.D., Katherine H. Walker, M.D., and Joseph
Loscalzo, M.D., Ph.D.
https://www.nejm.org/doi/full/10.1056/NEJMcps2304844?query=featured_home
Referencias
1. Baughman RP, Teirstein AS, Judson
MA, et al. Clinical characteristics
of patients
in a case control study of
sarcoidosis.
Am J Respir Crit Care Med 2001; 164:
1885-9.
2. Silverman KJ, Hutchins GM, Bulkley
BH. Cardiac sarcoid: a
clinicopathologic
study of 84 unselected patients with
systemic
sarcoidosis. Circulation 1978; 58:
1204-11.
3. Baughman RP, Field S, Costabel U,
et
al. Sarcoidosis in America: analysis
based
on health care use. Ann Am Thorac Soc
2016; 13: 1244-52.
4. Crouser ED, Maier LA, Wilson KC,
et
al. Diagnosis and detection of
sarcoidosis:
an official American Thoracic Society
clinical practice guideline. Am J
Respir Crit Care Med 2020; 201(8):
e26-
e51.
5. Chen ES, Moller DR. Sarcoidosis —
scientific progress and clinical
challenges.
Nat Rev Rheumatol 2011; 7: 457-67.
6. Huh J-Y, Moon DS, Song JW. Sarcoidlike
reaction in patients with malignant
tumors: long-term clinical course and
outcomes. Front Med (Lausanne) 2022;
9:
884386.
7. Kluger N. Tattoo-associated
uveitis
with or without systemic sarcoidosis:
a
comparative review of the literature.
J Eur
Acad Dermatol Venereol 2018; 32:
1852-61.
8. Haimovic A, Sanchez M, Judson MA,
Prystowsky S. Sarcoidosis: a
comprehensive
review and update for the
dermatologist:
part I. Cutaneous disease. J Am Acad
Dermatol 2012; 66(5): 699.e1-699.e18,
717-8.
9. Nishino M, Itoh H, Hatabu H. A
practical
approach to high-resolution CT of
diffuse lung disease. Eur J Radiol
2014; 83:
6-19.
10. Nakatsu M, Hatabu H, Morikawa K,
et
al. Large coalescent parenchymal
nodules
in pulmonary sarcoidosis: “sarcoid
galaxy”
sign. AJR Am J Roentgenol 2002; 178:
1389-93.
11. Jamilloux Y, Kodjikian L,
Broussolle
C, Sève P. Sarcoidosis and uveitis.
Autoimmun
Rev 2014; 13: 840-9.
12. James DG, Neville E, Siltzbach
LE. A
worldwide review of sarcoidosis. Ann
N Y
Acad Sci 1976; 278: 321-34.
13. Palmer DG, Schumacher HR.
Synovitis
with non-specific histological
changes
in synovium in chronic sarcoidosis.
Ann
Rheum Dis 1984; 43: 778-82.
14. Rahaghi FF, Baughman RP, Saketkoo
LA, et al. Delphi consensus
recommendations
for a treatment algorithm in
pulmonary
sarcoidosis. Eur Respir Rev 2020; 29:
190146.