Presentación de caso
Un hombre zurdo de 62 años fue atendido en la clínica de
trastornos de la memoria de este hospital debido a pérdida de memoria, cambios
de personalidad y comportamiento extraño.
Aproximadamente 5 años antes de esta evaluación, la esposa
del paciente notó que el paciente se estaba volviendo más olvidadizo y tomaba
siestas con frecuencia durante el día. También notó que le faltaba iniciativa
en su trabajo profesional; por ejemplo, no estaba cobrando ni pagando por
servicios. Los compañeros de trabajo del paciente observaron que tenía
dificultades para concentrarse y que con frecuencia requería reorientación al
interactuar con los clientes. El paciente no tenía más preocupaciones que dolores
de cabeza leves intermitentes.
Durante los años siguientes, la esposa del paciente notó que
se volvía distante con la familia, hablaba menos y carecía de interés en las
actividades que antes disfrutaba. Cometía numerosos errores en el trabajo y
también solía tener comportamientos extraños. Por ejemplo, entró a la cocina de
un restaurante local sin permiso; en otra ocasión, sin darse cuenta, pidió
prestado un vehículo de un colega sin notificarlo primero a esa persona. En
otro caso, mientras el vehículo del paciente estaba detenido por una infracción
de tráfico de rutina, se volvió irritable y eludió brevemente a la policía,
incurriendo en varias violaciones legales.
Un año antes de esta evaluación, el paciente fue atendido en
las clínicas de neurología y psiquiatría de otro hospital a pedido de su
esposa. No le preocupaba su estado neurocognitivo. Observó que dormía de 2 a 3
horas por noche, pero tenía un buen nivel de energía. Refirió dolores de cabeza
leves intermitentes. No tenía dolor en los senos nasales, entumecimiento,
hormigueo ni síntomas constitucionales. En el examen, el paciente estaba alerta
y orientado, con cierta falta de atención y confabulaciones. El resto del
examen neurológico fue normal. El hemograma completo y los niveles sanguíneos
de electrolitos y glucosa eran normales, al igual que los resultados de las
pruebas de función renal, hepática y tiroidea. Las pruebas de anticuerpos
antinucleares y de la enfermedad de Lyme fueron negativas. El paciente recibió
diagnósticos de trastorno por déficit de atención con hiperactividad, trastorno
de adaptación, y rasgos de personalidad evitativos. Se inició psicoterapia y se
prescribió melatonina y dextroanfetamina-anfetamina.
El paciente presentaba continuos cambios de personalidad y
dificultades de organización y funcionamiento diario, y fue evaluado en la
clínica de trastornos de la memoria de este hospital a solicitud de su esposa.
Informó que la dextroanfetamina-anfetamina no había mejorado sus capacidades
organizativas generales. Aunque era físicamente capaz de realizar actividades
de la vida diaria, necesitaba pautas para realizar tareas para mantener una
higiene básica, como bañarse. Ya no participó en la gestión de las finanzas
familiares; no pudo mantener el empleo y estaba recibiendo pagos por
discapacidad. Tenía nuevos comportamientos extraños, como orinar en público en
algunas ocasiones y visitar con frecuencia e inesperadamente los lugares de
trabajo de los miembros de la familia y las casas de los vecinos a altas horas
de la noche. También tenía comportamientos obsesivos, como llamar a sus hijos
varias veces para confirmar los detalles antes de una reunión programada. El
paciente desconocía sus comportamientos inapropiados e inusuales. No informó
tener ansiedad, depresión o alteraciones de la percepción.
Aproximadamente 10 años antes de la evaluación actual, el
paciente había recibido un diagnóstico de macroglobulinemia de Waldenström.
Debido a que estaba asintomático, el tratamiento se pospuso. Dos años antes de
la evaluación actual, se realizó una tomografía computarizada (TC) de tórax,
abdomen y pelvis para la vigilancia de la enfermedad. Existían múltiples
adenopatías hiliares y retrocrurales de hasta 1,2 cm en su mayor dimensión, sin
evidencia de organomegalia o lesiones focales óseas. El paciente acudió a
consultas de oncología de rutina para seguimiento de la enfermedad. Dos meses
antes de la evaluación actual, un hemograma completo y un panel metabólico
básico fueron normales. El nivel de IgM en sangre fue de 2850 mg por decilitro
(rango de referencia, 40 a 230) y el nivel de
β 2-microglobulina fue de 2.8 mg liter ( 0 to 2.7); esos valores habían
permanecido estables desde eldiagnóstico de macroglobulinemia de Waldenström.
Otros antecedentes médicos incluyeron insuficiencia mitral,
nefrolitiasis, hiperplasia prostática benigna, osteoartritis bilateral de rodilla
y estenosis espinal. No había antecedentes de traumatismo craneoencefálico,
accidente cerebrovascular o convulsiones. Los medicamentos incluían un
multivitamínico y aceite de pescado; no se conocían alergias a medicamentos. El
paciente no bebía alcohol, no fumaba tabaco ni consumía drogas ilícitas. Vivía
con su esposa y sus hijas y nietos vivían cerca. Se había graduado de la
escuela secundaria y la escuela comercial antes de servir en la marina durante
2 años; no sirvió en un papel de combate ni tuvo exposición conocida a toxinas.
Sus padres tenían una enfermedad cardíaca, una hermana tenía depresión y un tío
materno tenía cáncer de garganta. No había antecedentes familiares de demencia,
trastornos neurológicos o trastornos psiquiátricos.
En el examen, el paciente estaba bien arreglado, era
agradable y cooperaba. La apariencia física era normal. Su puntuación en el
Mini-Examen del Estado Mental fue de 23 en una escala que va de 0 a 30 (con
puntuaciones más altas indicando una mejor función cognitiva), y su puntuación
en la Evaluación Cognitiva de Montreal (MoCA) fue de 15 en una escala que va de
0 a 30 (con puntuaciones más altas indicando una mejor función cognitiva).
La orientación estaba intacta excepto por un error al
recordar la fecha. En la prueba de atención, el paciente tuvo dificultades con
los cálculos de siete en serie y con la prueba de intervalo de dígitos, en la
que el examinador lee una secuencia de números y el paciente la repite; repitió
sólo cuatro dígitos del tramo de avance correctamente. En la prueba de memoria,
repitió cinco de cinco palabras después de dos intentos y no recordó
espontáneamente ninguna de las palabras después de 5 minutos; recordó tres de
las palabras al seleccionar de una lista escrita de opciones de palabras. La producción
espontánea del habla estaba disminuida. En la prueba de nomenclatura de
confrontación, identificó un camello cuando se le mostró una imagen de una
jirafa. Al probar la fluidez semántica y fonémica, generó espontáneamente seis
nombres de animales durante un período de 1 minuto, así como una palabra que
comienza con la letra "F" durante un período de 1 minuto. La prueba
de dibujo del reloj mostró una buena organización. La prueba de secuenciación
de letras y números (también conocida como prueba de creación de senderos
alternos) reveló alteraciones en el cambio de conjuntos, y la prueba de tres
pasos de Luria reveló una alteración de la secuenciación motora. Al probar la
abstracción verbal, cuando se le pidió al paciente que describiera en qué se parecen
un tren y una bicicleta, afirmó: "Un tren es más grande que una bicicleta
y aplastaría la bicicleta". El juicio y la perspicacia se vieron
notablemente afectados. Cuando se le pidió al paciente que describiera en qué
se parecen un tren y una bicicleta, dijo: "Un tren es más grande que una
bicicleta y aplastaría la bicicleta". El juicio y la perspicacia se vieron
notablemente afectados.
Una evaluación de la función, sensibilidad, fuerza, volumen,
tono, coordinación y marcha de los pares craneales fue normal. Los reflejos
tendinosos profundos eran normales y los dedos de los pies descendían
bilateralmente. Estaban presentes reflejos bilaterales de prensión y reflejos
palmomentonianos. La prueba de sífilis fue negativa. El nivel de folato en
sangre fue de 8.2 ng por mililitro (más de 4.7 ng por mililitro), y el nivel de
vitamina B 12 258 pg por mililitro (más de 231 pg por mililitro).
En la evaluación neuropsicológica, el compromiso social fue
limitado, con algunos momentos de risitas que eran incongruentes con el
contexto. El habla espontánea era fluida pero empobrecida. Hubo un uso
frecuente de la palabra "cosa" para reemplazar palabras de menor
frecuencia y el uso periódico de parafasias fonémicas. Los pensamientos eran
coherentes pero empobrecidos, tangenciales y perseverantes. La atención y el
funcionamiento ejecutivo se vieron marcadamente afectados durante las
evaluaciones de la fluidez verbal, la atención dividida, el razonamiento
abstracto y el control inhibitorio. La memoria visual estaba relativamente
intacta, pero la memoria verbal estaba deteriorada en términos de codificación
y recuperación. Cuando se le pidió al paciente que describiera una imagen, el
lenguaje escrito se vio gravemente afectado, pero el lenguaje oral estaba
relativamente intacto.
Se revisó una prueba de diagnóstico y se realizó un
diagnóstico.
Diagnóstico diferencial
Este hombre de 62 años tuvo una pérdida de memoria
clínicamente significativa y cambios de personalidad y de comportamiento que
deterioraron progresivamente su capacidad para funcionar profesional y
personalmente durante el transcurso de 5 años. Las características de su
presentación se ajustan a los criterios para la demencia caracterizada por un
inicio insidioso con progresión en el tiempo. Desarrollaré un diagnóstico
diferencial de demencia que se centra en la intersección de enfermedad
neurológica y psiquiátrica. 1
EVALUACIÓN DE HALLAZGOS
Los hallazgos iniciales observados en este paciente fueron
olvido, dificultad para concentrarse, sueño interrumpido, apatía y falta de
preocupación seguida de confabulación. La alteración del ritmo circadiano que
conduce a la alteración o la pérdida del sueño nocturno se asocia cada vez más
con la demencia temprana. 2 La apatía implica una disminución o pérdida de la
motivación, en comparación con el nivel anterior de funcionamiento,
característica que no concuerda con la edad o cultura del paciente. 3 La
confabulación espontánea, definida como la generación de narrativas falsas con
ausencia de duda en la formación y expresión de las creencias, 4 se asocia
principalmente con la demencia amnésica, pero también se puede ver con
neurosífilis, hipoxemia, trastornos del pensamiento y psicosis afectivas.
La inexorable progresión de la enfermedad del paciente
provocó una franca desinhibición e impulsividad, así como conductas
compulsivas. 5 Cuatro años después de iniciado el curso de la enfermedad,
recibió diagnósticos psiquiátricos de trastorno por déficit de atención con
hiperactividad, trastorno de adaptación y rasgos de personalidad por evitación.
Eventualmente se volvió incapaz de trabajar y ya no estuvo involucrado en las
decisiones familiares. Tuvo una disminución en su higiene personal, una falta
de conciencia de sus deficiencias y comportamientos inusuales (incluido el robo
inadvertido y la micción en público), y resultados normales en exámenes
neurológicos elementales seriados, excepto por la presencia de reflejos de
prensión y reflejos palmomentales, que son Hallazgos inespecíficos. 5,6
EVALUACIÓN COGNITIVA DE MONTREAL
Cinco años en el curso de la enfermedad, el paciente se
sometió a la MoCA, que fue esclarecedor. 7La atención, la motivación y el
esfuerzo son cruciales para establecer la validez de la evaluación. El proceso,
no solo la puntuación final, es importante para la interpretación de los
resultados. El paciente no pudo completar con éxito la prueba de secuenciación
de letras y números. No hizo ningún esfuerzo por autocorregirse, lo que implica
apatía y falta de conciencia. Tuvo una leve falta de atención, fue capaz de
recitar solo cuatro dígitos del intervalo hacia adelante en la prueba de
intervalo de dígitos y generó solo 1 palabra que comienza con la letra
"F" durante un período de 1 minuto, mientras que los graduados de la
escuela secundaria normalmente generan 11 palabras o más. La abstracción verbal
se interpretó de forma concreta. En la prueba de la memoria verbal, el paciente
aprendió 5 palabras durante una segunda prueba y espontáneamente no recordó
ninguna de las palabras después de un tiempo. Sin embargo, cuando le dieron
múltiples opciones escritas, recordó correctamente 3 de las palabras, un
hallazgo que sugiere cierta retención de memoria. Una interpretación plausible
es que los resultados de MoCA no sugieren amnesia primaria, sino que reflejan
amnesia secundaria debido a un estado apático, desconectado y despreocupado.
Una evaluación neuropsicológica formal realizada 5 años después del inicio de
la enfermedad reveló un compromiso limitado con la prueba, un afecto plano,
risitas incongruentes, habla y lenguaje fluidos con contenido empobrecido y
errores parafásicos, atención y funcionamiento ejecutivo notablemente
deteriorados y memoria verbal deteriorada con efectos de memoria visuales relativamente intactos.
HISTORIAL DEL PACIENTE
Se obtuvo una historia relevante del paciente, con un
enfoque en el tiempo anterior al inicio de la enfermedad. El paciente se había
graduado de la escuela secundaria y la escuela comercial, lo que sugiere que
tenía un funcionamiento cognitivo inicial normal. Había recibido un diagnóstico
de macroglobulinemia de Waldenström, que se describió como estable sin
tratamiento. El linfoma linfoplasmocítico puede causar una enfermedad del
sistema nervioso central que produce deterioro cognitivo y síntomas
psiquiátricos, aunque esta manifestación es rara. 8Sin embargo, este
diagnóstico suele ir acompañado de déficits motores y parálisis de pares
craneales, que no estaban presentes en este paciente. Además, una tomografía
computarizada de tórax de vigilancia obtenida 2 años antes de la evaluación
actual no mostró evidencia de organomegalia o lesiones óseas focales; estos
resultados reducen la probabilidad de cáncer o compromiso cerebral
paraneoplásico.
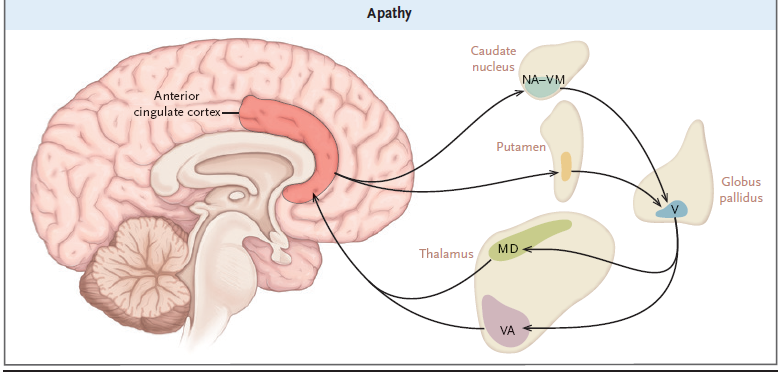
DISFUNCIÓN DE LA RED FRONTAL
Anatomía del cerebro asociada con los primeros síntomas neuroconductuales
(neuropsiquiátricos) de este paciente.
Muchas afecciones neurológicas comunes se asocian con la
disfunción de la red frontal, que causa cambios clínicamente significativos en
la personalidad y el comportamiento. Los circuitos frontales subcorticales
transportan proyecciones desde los tálamos hasta los lóbulos frontales y
conectan los lóbulos frontales con los ganglios basales. Las alteraciones en
los circuitos frontales-subcorticales no motores están específicamente
relacionadas con la apatía, la desinhibición y la disfunción ejecutiva,
características destacadas de la presentación de este paciente ( Figura 1 ).
Figura 1. Anatomía
cerebral asociada con síntomas neuroconductuales (neuropsiquiátricos)
tempranos.
El cerebro está compuesto de regiones anatómicas con
propiedades arquitecturales únicas que están distribuidas geográficamente a
todo lo largo de las áreas corticales y subcorticales y atadas con precisión
topográfica.
Significado de las siglas:
DL denotes dorsolateral, LDM dorsomedial lateral, MD mediodorsal, MDM
dorsomedial medial, NA nucleus accumbens, V ventral, VA ventral anterior, and
VM ventromedial.
CAUSAS DE DEMENCIA ASOCIADA CON LESIONES CEREBRALES
Los resultados normales de este paciente en los exámenes
neurológicos seriados no serían típicamente consistentes con los diagnósticos
asociados con lesiones cerebrales. Sin embargo, las presentaciones atípicas no
son infrecuentes con estos diagnósticos, y el juicio clínico sigue siendo
fundamental para descartarlos. Uno de esos diagnósticos es la enfermedad
vascular cerebral; el paciente no tenía factores de riesgo vascular conocidos,
aunque sería importante conocer su índice de masa corporal y determinar si
padecía apnea obstructiva del sueño. La enfermedad de Huntington es otra posibilidad,
dado que su pico de incidencia se da en la cuarta y quinta décadas de la vida,
pero este paciente, ahora en su séptima década de vida, no tenía antecedentes
familiares de enfermedad de Huntington y no había reportado trastornos del movimiento durante 5 años en el
curso de la enfermedad. 9
La demencia con cuerpos de Lewy es un diagnóstico poco
probable en este paciente porque no tenía características parkinsonianas,
inestabilidad postural, disfunción autonómica, caídas repetidas, delirios o alucinaciones
visuales. Las taupatías, como la parálisis supranuclear progresiva, pueden
descartarse sobre la base de los resultados normales de los exámenes
neurológicos seriados, incluidos los movimientos extraoculares intactos, que se
observaron cinco años después de la evolución de la enfermedad. La enfermedad
de la sustancia blanca, como la esclerosis múltiple, es una posibilidad, pero
no tenía ninguno de los signos o síntomas típicos. También se deben considerar
los tumores de crecimiento lento, incluidos los meningiomas que emanan de la
hoz del cerebro o la placa cribiforme y los tumores hipofisarios que invaden
las estructuras supraselares, pero son poco probables.
La presentación clínica del paciente podría ser compatible
con hidrocefalia normotensiva, pero su marcha normal y la ausencia de
incontinencia no son compatibles con este diagnóstico. Las infecciones comunes,
como la infección por el virus de la inmunodeficiencia humana y la sífilis,
siempre deben considerarse en el diagnóstico diferencial del deterioro
neurocognitivo progresivo, pero las pruebas de sífilis fueron negativas, no
tenía factores de riesgo de infección y su examen neurológico no presentaba
complicaciones. Las enfermedades priónicas, enfermedad de Creutzfeldt-Jakob
particularmente variante, 10 deben ser considerados en este paciente, aunque la
edad media al inicio de la enfermedad es de 29 años; además, no tenía
mioclonías, características piramidales o extrapiramidales, ni signos
cerebelosos y había sobrevivido mucho más tiempo de lo esperado con esta
enfermedad.
La hipotensión intracraneal espontánea puede causar síndrome
de flacidez cerebral frontotemporal 11 con desconexión frontal-subcortical,
pero los síntomas suelen depender de la posición del paciente. La encefalopatía
de Hashimoto también debe considerarse en este caso, pero los resultados de las
pruebas de laboratorio fueron normales y no hubo hallazgos físicos compatibles
con enfermedad de la tiroides, como temblores, convulsiones, mioclonías o
ataxia.
VARIANTE CONDUCTUAL DE LA DEMENCIA FRONTOTEMPORAL
Los dos diagnósticos más probables en este caso son la
variante conductual de la demencia frontotemporal (bvFTD) 12 y la variante
frontal de la enfermedad de Alzheimer (fvAD). 13 Puede ser difícil distinguir
la fvAD con rasgos frontales desproporcionados de la bvFTD. La aparición de una
prominente pérdida de memoria temprana con bvFTD puede desdibujar aún más la
distinción. Este paciente tenía cinco de los seis síntomas principales de bvFTD
( Tabla 1 ): desinhibición conductual temprana, apatía, pérdida de simpatía o
empatía, conductas compulsivas o ritualistas y déficits en el funcionamiento
ejecutivo con relativa conservación de la memoria y las habilidades
visuoespaciales. 14Es probable que se produzcan hiperoralidad (masticación
excesiva, succión o relamerse los labios) y los cambios asociados con la
preferencia de alimentos o la dieta (p. Ej., atracones, aumento de peso o
ingesta de objetos no comestibles) en pacientes con bvFTD, pero no se
informaron tales cambios en esta caso. Los pacientes con bvFTD a menudo se
derivan para una evaluación psiquiátrica y pueden tener resultados normales en las
neuroimágenes al principio del curso de la enfermedad; la enfermedad puede
manifestarse de forma simétrica o asimétrica, tanto clínica como en las
neuroimágenes.


Tabla 1.
Criterios de diagnóstico para la variante conductual de la
demencia frontotemporal.
Varias características de la presentación de este paciente
favorecen el diagnóstico de bvFTD. Tenía 57 años al inicio de la enfermedad; la
frecuencia de aparición temprana de la enfermedad (menos de 65 años de edad)
entre los pacientes con bvFTD es similar o mayor que la frecuencia entre
aquellos con fvAD. Tuvo cambios profundos tempranos en la personalidad y el
comportamiento. La apatía es el síntoma inicial más común de bvFTD y es más
frecuente y grave entre los pacientes con bvFTD que entre aquellos con fvAD. 16
Los comportamientos delictivos, como el robo, son más comunes entre los pacientes
con bvFTD.La puntuación MoCA de este paciente, que indicó una disfunción
ejecutiva y conductual mayor posiblemente sin amnesia primaria, y su perfil
neuropsicológico asimétrico, que indicaba una memoria verbal deteriorada con
una memoria visual relativamente intacta, también favorecen el diagnóstico de
bvFTD. El síndrome clínico de bvFTD implica una neuroanatomía selectiva, pero
puede no ser predictivo de las características patológicas subyacentes
precisas. Para establecer el diagnóstico de bvFTD, recomendaría una resonancia
magnética (MRI) de la cabeza para evaluar la atrofia de los lóbulos
frontotemporales.
DIAGNÓSTICO PRESUNTIVO
VARIANTE CONDUCTUAL DE LA DEMENCIA FRONTOTEMPORAL.
La resonancia magnética de la cabeza, realizada antes y
después de la administración de contraste intravenoso ( Figura 2 ), reveló una
pérdida de volumen desproporcionada que era más prominente en los lóbulos
frontal y temporal y mayor en el lado izquierdo que en el derecho. , con
afectación de las circunvoluciones del cíngulo anterior y dilatación ex vacuo
de los cuernos frontales y temporales adyacentes de los ventrículos laterales. No
había infarto cerebral, masa intracraneal, realce intracraneal anormal o
evidencia de hemorragia previa. Este patrón de pérdida de volumen
parenquimatoso fue compatible con el diagnóstico clínico de demencia
frontotemporal.
Figura 2. Resonancia magnética de la cabeza.
Una imagen en FLAIR (Panel A) muestra una pérdida de volumen
desproporcionada, que es más prominente en los surcos corticales a lo largo de la
parte anterior y medial del lóbulo
frontal izquierdo (puntas de flecha) que en los surcos parietal y occipital.
Una imagen axial en FLAIR a través de
los lóbulos frontal y temporal (Paneles B y C, respectivamente) muestran pérdida
de volumen asimétrica, que es más prominente en el lado izquierdo (flechas) que
en el lado derecho.
DISCUSIÓN DEL MANEJO
Sobre la base de la presentación clínica de este paciente y
la atrofia de predominio frontotemporal en la resonancia magnética, la
estrategia de manejo inicial se centró en brindar asesoramiento y apoyo al
paciente y su esposa. 12 La asesoría incluyó una revisión de los síntomas
centrales y la historia natural de bvFTD (incluida la desinhibición, apatía,
pérdida de empatía, desarrollo de comportamientos estereotipados o ritualistas,
hiperoralidad, cambios en la dieta y disfunción ejecutiva), 14 así como una
discusión de los sorprendentes pérdida de conocimiento que comúnmente acompaña
a esta condición. 17Como es una práctica común en el tratamiento de
enfermedades neurodegenerativas para las que aún no se dispone de terapias
modificadoras de la enfermedad, se programaron visitas clínicas de seguimiento
para pacientes ambulatorios para brindar apoyo emocional continuo a la esposa
del paciente y otros miembros de la familia. También se revisaron los recursos
en línea disponibles y los servicios de trabajo social.
Para el manejo farmacológico, se recetó sertralina, un
inhibidor selectivo de la recaptación de serotonina, para atacar la
impulsividad y las conductas compulsivas emergentes. 18 Se recetó trazodona
para aliviar el insomnio. Durante varias visitas de seguimiento, se discutieron
estrategias de manejo conductual no farmacológico con la esposa del paciente.
19
Durante los 3 años posteriores a la visita inicial del
paciente, su condición neurocognitiva continuó disminuyendo. Comenzó a tener
conductas motoras repetitivas, incluido el golpeteo frecuente de las rodillas y
el rechinar de dientes. 14 También surgieron patrones inusuales de
alimentación; en una ocasión comió posos de café y se obsesionó con beber una
marca de café en particular. La esposa del paciente y otros miembros de la
familia notaron que tenía una marcada pérdida de empatía. Hubo una disminución
clínicamente significativa en la producción del habla; a veces, respondía a las
preguntas con solo "sí" o "no", y después de la progresión
posterior, respondía con "ajá". Fue remitido a un programa diurno
para adultos, que, según los informes, fue una experiencia positiva para el
paciente. A los 66 años, requirió atención completa y ya no regresó a la
clínica. Murió a los 69 años. Con el permiso de su familia, se realizó una
autopsia.
DISCUSIÓN PATOLÓGICA
Se realizó una autopsia que se limitó al examen del cerebro.
En el examen macroscópico ( Figura 3), el cerebro pesaba 910 g (rango normal,
1250 a 1400) y tenía atrofia severa de los lóbulos frontales y la porción
anterior de los lóbulos temporales que era mayor en el lado izquierdo que en el
derecho. Había un adelgazamiento cortical severo en las áreas de atrofia y
separación focal de la banda cortical de la sustancia blanca subyacente, que
estaba decolorada y firme. Había una notable ausencia de atrofia en la porción
posterior de la circunvolución temporal superior. La amígdala y el hipocampo
estaban levemente atróficos. El resto del examen general del cerebro no tuvo
nada de especial.
Figura 3. Fotografías del cerebro en la autopsia.
La superficie inferior del cerebro (Panel A) muestra atrofia
de los lóbulos temporales frontal y anterior que es mayor en el lado izquierdo
que en el derecho. Secciones coronales del lóbulo frontal anterior y del lóbulo
temporal anterior (Panel B) muestran atrofia severa, junto con dilatación
ventricular. Se muestran secciones coronales más posteriores (Panel C) con relativa
preservación de la circunvolución temporal superior (puntas de flecha) y partes
posteriores del lóbulo frontal.
El examen microscópico ( Figura 4 ) reveló una corteza
severamente degenerada en las áreas del cerebro muy afectadas, con estado de
espongiosis, astrogliosis y pérdida neuronal casi completa. Había inclusiones
citoplasmáticas eosinofílicas redondeadas en muchas neuronas de la
circunvolución dentada del hipocampo y dispersas por otras áreas corticales.
Había neuronas hinchadas ocasionales. También se presentó enfermedad
cerebrovascular, con aterosclerosis leve de las arterias carótidas internas y
arteriolosclerosis leve de vasos en las leptomeninges y sustancia blanca.
Figura 4. Muestras de cerebro en autopsia.
Una tinción con luxol con azul rápido con hematoxilina y eosina de
la corteza frontal inferior a bajo y alto aumento (Paneles A y B,
respectivamente) muestra el estado de la espongiosis, astrogliosis y pérdida
neuronal en la corteza. Allí también hay pérdida de tinción de mielina azul
rápido Luxol en la materia blanca subyacente debido a la degeneración axonal (Panel
A). Es visible una rara neurona cortical restante (Panel B, punta de flecha).
Luxol fast blue-hematoxilina y tinción con eosina de la circunvolución dentada
del hipocampo en gran aumento (Panel C) muestra numerosos inclusiones redondeadas
citoplasmáticas eosinofílicas, conocidas como cuerpos de Pick (puntas de flecha). Sobre tinción
inmunohistoquímica de la circunvolución dentada del hipocampo (Panel D), los
cuerpos de Pick son positivos para tau (en marrón). La tinción inmunohistoquímica
de la circunvolución parahipocampal con bajo aumento y alto aumento (paneles E y F, respectivamente) muestra
numerosos cuerpos Pick redondeados con tau positivo y neuritas distróficas
(Panel F, punta de flecha). La tinción inmunohistoquímica de la circunvolución
del cíngulo en gran aumento (Panel G)
muestra neuronas hinchadas tau positivo, conocidas como células Pick (puntas de
flecha), así como cuerpos Pick redondeados tau-positivos.
El patrón de atrofia macroscópica sugiere fuertemente un
diagnóstico subyacente de degeneración lobar frontotemporal (FTLD)( frontotemporal
lobar degeneration). La atrofia fue más restringida de lo que cabría esperar en
un paciente con enfermedad de Alzheimer y no permaneció dentro de los
territorios vasculares, como cabría esperar en un paciente con lesión
isquémica. Los hallazgos macroscópicos están totalmente respaldados por los
cambios microscópicos en las regiones corticales afectadas.
La FTLD se caracteriza por agregados de proteínas
ubiquitinadas, más comúnmente en neuronas. Los subtipos se basan en la
proteinopatía específica e incluyen FTLD-tau (con inclusiones tau positivas;
las subcategorías incluyen enfermedad de Pick, mutación MAPT y otras), FTLD-TDP
(con inclusiones positivas para la proteína de unión al ADN TAR 43), FTLD-FUS
(con inclusiones positivas para la proteína fusionada en sarcoma), FTLD-UPS
(con inclusiones positivas para los marcadores del sistema del proteasoma de
ubiquitina) y FTLD-ni (sin inclusiones). 21 Para determinar el subtipo de la
enfermedad de este paciente se realizó tinción inmunohistoquímica ( Figura 4).
La tinción reveló numerosas inclusiones intraneuronales redondeadas positivas
para tau (cuerpos de Pick) en regiones corticales muy afectadas, incluida la
corteza frontal, la circunvolución dentada del hipocampo y la circunvolución
parahipocampal. También se identificaron neuronas tau positivas inflamadas
(células Pick) y neuritas distróficas. La presencia de cuerpos de Pick
positivos para tau y células de Pick es un diagnóstico de la enfermedad de
Pick, una subcategoría de FTLD-tau que se caracteriza por la agregación de
isoformas de tau 3R. Las tinciones adicionales para TDP-43 fosforilado y
α-sinucleína fueron negativas. Una tinción para β-amiloide reveló placas
difusas raras en el hipocampo y la neocorteza, un hallazgo compatible con el
envejecimiento normal. Sobre la base de las características clínicas,
radiológicas e histológicas identificadas en este paciente, el diagnóstico
final es FTLD compatible con la enfermedad de Pick.
DIAGNOSTICO FINAL
DEGENERACIÓN LOBAR FRONTOTEMPORAL CON INCLUSIONES
TAU-POSITIVAS (FTLD-TAU) COMPATIBLE CON LA ENFERMEDAD DE PICK.
Traducción
de:
A
62-Year-Old Man with Memory Loss and Odd Behavior
Bruce H. Price, M.D., David L. Perez, M.D., M.M.Sc., Otto
Rapalino, M.D., and Derek H. Oakley, M.D., Ph.D.
NEJM
https://www.nejm.org/doi/10.1056/NEJMcpc1916251
References
1.
Arvanitakis Z, Shah RC, Bennett DA.
Diagnosis
and management of dementia:
review.
JAMA 2019; 322: 1589-99.
2. Musiek
ES, Holtzman DM. Mechanisms
linking
circadian clocks, sleep, and
neurodegeneration.
Science 2016; 354:
1004-8.
3. Cummings
JL. Toward a molecular
neuropsychiatry
of neurodegenerative diseases.
Ann Neurol
2003; 54: 147-54.
4. McKay R,
Kinsbourne M. Confabulation,
delusion,
and anosognosia: motivational
factors and
false claims. Cogn
Neuropsychiatry
2010; 15: 288-318.
5. Mendez
MF, Chen AK, Shapira JS,
Miller BL.
Acquired sociopathy and frontotemporal
dementia.
Dement Geriatr Cogn
Disord
2005; 20: 99-104.
6. Thomas
RJ. Blinking and the release
reflexes: are
they clinically useful? J Am
Geriatr Soc
1994; 42: 609-13.
7.
Nasreddine ZS, Phillips NA, Bédirian
V, et al.
The Montreal Cognitive Assessment,
MoCA: a
brief screening tool for
mild
cognitive impairment. J Am Geriatr
Soc 2005;
53: 695-9.
8. Malkani
RG, Tallman M, Gottardi-
Littell N,
et al. Bing-Neel syndrome: an
illustrative
case and a comprehensive review
of the
published literature. J Neurooncol
2010; 96:
301-12.
9. Walker
FO. Huntington’s disease.
Lancet
2007; 369: 218-28.
10. Appleby
BS, Appleby KK, Crain BJ,
Onyike CU,
Wallin MT, Rabins PV. Characteristics
of
established and proposed
sporadic
Creutzfeldt-Jakob disease variants.
Arch Neurol
2009; 66: 208-15.
11.
Wicklund MR, Mokri B, Drubach DA,
Boeve BF,
Parisi JE, Josephs KA. Frontotemporal
brain
sagging syndrome: an
SIH-like
presentation mimicking FTD.
Neurology
2011; 76: 1377-82.
12. Seeley
WW. Behavioral variant frontotemporal
dementia.
Continuum (Minneap
Minn) 2019;
25: 76-100.
13.
Ossenkoppele R, Pijnenburg YAL, Perry
DC, et al.
The behavioural/dysexecutive
variant of
Alzheimer’s disease: clinical,
neuroimaging
and pathological features.
Brain 2015;
138: 2732-49.
14.
Rascovsky K, Hodges JR, Knopman
D, et al.
Sensitivity of revised diagnostic
criteria
for the behavioural variant of
frontotemporal
dementia. Brain 2011;
134:
2456-77.
15.
Ducharme S, Price BH, Larvie M,
Dougherty
DD, Dickerson BC. Clinical approach
to the
differential diagnosis between
behavioral
variant frontotemporal
dementia
and primary psychiatric disorders.
Am J
Psychiatry 2015; 172: 827-37.
16.
Ducharme S, Price BH, Dickerson BC.
Apathy: a
neurocircuitry model based on
frontotemporal
dementia. J Neurol Neurosurg
Psychiatry
2018; 89: 389-96.
17. Banks
SJ, Weintraub S. Generalized
and
symptom-specific insight in behavioral
variant
frontotemporal dementia
and primary
progressive aphasia. J Neuropsychiatry
Clin
Neurosci 2009; 21: 299-
306.
18. Swartz
JR, Miller BL, Lesser IM, Darby
AL.
Frontotemporal dementia: treatment
response to
serotonin selective reuptake
inhibitors.
J Clin Psychiatry 1997; 58: 212-
6.
19. Barton
C, Ketelle R, Merrilees J, Miller
B.
Non-pharmacological management of
behavioral
symptoms in frontotemporal
and other
dementias. Curr Neurol Neurosci
Rep 2016;
16: 14.
20.
Grinberg A, Lagunoff J, Phillips D,
Stern B,
Goodman M, Chow T. Multidisciplinary
design and
implementation of a
day program
specialized for the frontotemporal
dementias.
Am J Alzheimers Dis
Other Demen
2007; 22: 499-506.
21. Kalaria
JL. Dementia. In: Love S, Perry
A, Ironside
J, Budka H, eds. Greenfield’s
neuropathology. Boca Raton, FL: CRC
Press, 2015: 858-973.