La mayoría de las enfermedades reumáticas pueden conceptualizarse como trastornos inflamatorios o no inflamatorios. Las enfermedades autoinmunes idiopáticas comprenden la mayoría de los trastornos reumáticos inflamatorios. Algunos trastornos inflamatorios tienen la fibrosis como manifestación principal. Los trastornos inflamatorios con un origen mejor definido incluyen las enfermedades microcristalinas (p. ej., gota y seudogota) y las muchas artritis infecciosas. Las pruebas de laboratorio y los estudios de imagen en reumatología generalmente se dividen en dos categorías: los que se usan para ayudar al diagnóstico y los que se usan para monitorear la terapia. La mayor parte de la farmacoterapia en enfermedades reumáticas está dirigida contra la inflamación, el dolor o ambos.
TRASTORNOS AUTOINMUNE IDIOPATICOS
ARTRITIS REUMATOIDE
La artritis reumatoide (AR) es una afección autoinmune
sistémica que causa principalmente inflamación del tejido sinovial (sinovitis)
con la consiguiente artritis inflamatoria y tenosinovitis. La AR clásicamente
se presenta como una poliartritis simétrica de las pequeñas articulaciones de
las extremidades superiores e inferiores, aunque casi cualquier articulación
puede estar involucrada. La AR se presenta con mayor frecuencia en pacientes
con un inicio máximo entre los 25 y los 55 años de edad y una proporción de 2:1
entre mujeres y hombres. Sin embargo, todas las edades, géneros y razas pueden
verse afectados.
El proceso patológico característico es la sinovitis
proliferativa, que puede conducir a la destrucción del cartílago, erosión ósea
y artritis deformante. Los pacientes con AR también pueden experimentar
síntomas extraarticulares, más comúnmente síntomas constitucionales y secos,
nódulos cutáneos, enfermedad pulmonar intersticial (EPI), serositis (efusiones
pleurales y pericárdicas), anemia y trombocitosis. Las complicaciones menos
comunes incluyen vasculitis, amiloidosis secundaria, enfermedad ocular,
síndrome de Felty, síndrome de linfocitos granulares grandes y linfomas (Tabla
23.2) para una lista completa de complicaciones extraarticulares de la AR.
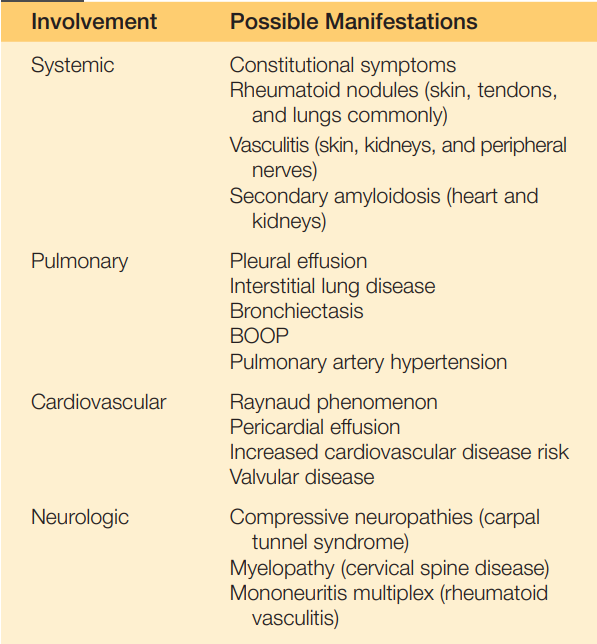

Tabla 23.2. Tabla 23.2. Manifestaciones extraarticulares de la artritis reumatoidea (no incluye potenciales toxicidades medicamentosas).
El diagnóstico de la AR se ayuda con la prueba del
factor reumatoideo (RF) sérico y los anticuerpos contra el péptido citrulinado
cíclico (CCP) (ACPA). Estos están presentes en aproximadamente el 80 % de los
pacientes con AR, siendo las pruebas ACPA de anticuerpos anti-CCP mucho más
específicas para la AR que el RF en suero. Es importante destacar que alrededor
del 20% de los pacientes con AR nunca muestran valores anormales de RF o
anti-CCP ACPA. Los marcadores de laboratorio de inflamación (Cuadro 30.1) no
son específicos, pero a menudo se correlacionan con la actividad de la enfermedad.
El análisis del líquido sinovial en un paciente con AR activa demuestra
>2000 glóbulos blancos (WBC)/mm3, pero este hallazgo no es específico y no
es diagnóstico de AR. La artrocentesis en este contexto se usa principalmente
para establecer si el derrame es inflamatorio o no y para excluir otros
diagnósticos (p. ej., artritis séptica o microcristalina). La radiografía
simple puede mostrar inflamación de los tejidos blandos, osteopenia
periarticular, estrechamiento del espacio articular y erosiones marginales. La
resonancia magnética y la ecografía musculoesquelética están desempeñando un
papel cada vez más importante en el diagnóstico de la AR.

Históricamente, los fármacos antiinflamatorios no
esteroideos (AINE) y los analgésicos fueron los pilares del tratamiento de la
AR. Sin embargo, más recientemente se han logrado grandes avances en el
tratamiento de la AR. Estos tratamientos se enfocan en interrumpir la sinovitis
proliferativa inflamatoria. Los corticosteroides han logrado esto, pero con
muchos efectos secundarios potenciales (Cuadro 30.2).

Cuadro 30.2. Efectos secundarios de los
corticosteroides sistémicos
Las estrategias de tratamiento más recientes tienen
como objetivo limitar el uso de corticosteroides y afectar la progresión de la
enfermedad utilizando fármacos antirreumáticos modificadores de la enfermedad
(DMARD). Para obtener una lista de DMARD, sus mecanismos de acción y perfiles
de efectos secundarios, consulte la Tabla 23.3.


Tabla 23.3 Lista de agentes farmacológicos utilizados
para el tratamiento de la artritis reumatoide
Los ejemplos de DMARD tradicionales incluyen
metotrexato, leflunomida, hidroxicloroquina, sulfasalazina, oro y muchos otros.
La última década ha sido testigo del advenimiento de la terapia biológica
DMARD, con fármacos que antagonizan el factor de necrosis tumoral-α (TNF-α)
(adalimumab, certolizumab, etanercept, golimumab, infliximab y versiones
biosimilares de infliximab), bloquean la interleucina (IL )-6 del receptor de
señalización (tocilizumab y sarilumab), bloquean la coestimulación alterada de
las células T (abatacept) o causan el agotamiento de las células B (rituximab).
Las terapias emergentes incluyen el tofacitinib, un inhibidor de la cinasa de
Janus de molécula pequeña. Colectivamente, estas nuevas clases de medicamentos,
aunque son agentes inmunosupresores potentes, han revolucionado el tratamiento
de la AR, creando un cambio de paradigma en los objetivos del tratamiento que se
alejan de la paliación y se acercan a la remisión.
POLIMIALGIA REUMÁTICA
La polimialgia reumática (PMR) es un trastorno
inflamatorio sistémico de personas mayores de 50 años. Por lo general, se
presenta como dolor en la cintura de los miembros de los hombros y las caderas
fuera de proporción con los hallazgos del examen, con una preponderancia de
rigidez matutina. La presentación suele ser de inicio súbito y en ocasiones se
asocia con sinovitis de las pequeñas articulaciones periféricas. Las pruebas de
laboratorio muestran evidencia de inflamación sistémica (ver Cuadro 30.1). La
tasa de sedimentación de eritrocitos (VSG) puede ser >100 mm por hora. Pocos
otros estados de enfermedad se asocian con una VSG tan elevada, lo cual es un
hallazgo poco común (Cuadro 30.3). Hasta el 5% al 10% de los pacientes con
PMR evolucionarán a una enfermedad parecida a la AR. Hasta el 15% al 20% de
los pacientes con PMR desarrollarán síntomas de arteritis de células gigantes
(discutido más adelante). Los corticosteroides en dosis bajas (típicamente 15 a
20 mg diarios) son el tratamiento inicial primario para la PMR. Los esteroides
se reducen según lo permitan los síntomas, a veces durante 1 o 2 años.

Cuadro 30.3 Diagnóstico diferencial de enfermedades asociadas con una tasa de sedimentación globular >100 mm/h.
ESPONDILOARTRITIS
Las espondiloartritis (SPA) son una colección de trastornos
que incluyen la espondilitis anquilosante, la artritis psoriásica, la
artropatía asociada con la enfermedad inflamatoria intestinal (artropatía por
EII), la artritis posinfecciosa (reactiva) y la espondiloartropatía no
diferenciada. A diferencia de la AR, la artritis periférica es típicamente
oligoarticular, asimétrica y más pronunciada en las extremidades inferiores.
Otras características distintivas de los SPA incluyen inflamación lumbosacra
(espondilitis o sacroilitis), entesitis y lesiones formadoras de hueso por
radiografía. La rigidez matutina de la espalda en una persona joven debe
alertar al médico sobre la posibilidad de una enfermedad inflamatoria de la
espalda.
La espondilitis anquilosante (EA) tiene un predominio
de >3:1 de hombre a mujer. AS afecta principalmente a la columna vertebral y
puede conducir a la fusión (anquilosis) de las vértebras. Las mujeres pueden
desarrollar EA de forma atípica con afectación del cuello antes que la columna
vertebral y la articulación sacroilíaca. La artritis psoriásica y la artropatía
por EII se asocian con sus respectivas entidades patológicas, pero la
afectación de la columna puede causar "áreas salteadas" en lugar de
una progresión uniforme como se observa en la EA. La artritis puede preceder a
la enfermedad de la piel o del intestino en estos trastornos. La artritis
reactiva se asocia con antecedentes de infección por clamidia o patógenos
intestinales enteroinvasivos (shigella, salmonella o yersinia). Las
manifestaciones extraarticulares adicionales de todas las SPA pueden incluir
dactilitis (dedo en salchicha), uveítis, balanitis circinada y lesiones
plantares escamosas (queratodermia blenorrágica). En raras ocasiones, estas
condiciones se asocian con aneurismas aórticos proximales y fibrosis pulmonar.
El estudio de laboratorio de los SPA no revela
anticuerpos antinucleares (ANA), FR o ACPA. Aunque las pruebas de laboratorio
no son específicas, pueden mostrar inflamación sistémica (ver cuadro 30.1) y
los glóbulos blancos del líquido sinovial pueden estar extremadamente elevados.
La afectación espinal está estrechamente asociada con el antígeno leucocitario
humano (HLA)-B27. La utilidad diagnóstica de la prueba del estado HLA es
discutible, aunque los criterios más nuevos incorporan esta prueba. La entesitis,
la anquilosis y la sacroilitis son hallazgos radiográficos distintivos. El
tratamiento de las SPA refleja fielmente el de la AR. Es posible que las
terapias tradicionales con DMARD (incluidos el metotrexato y la sulfasalazina)
solo tengan un papel en el tratamiento de la SPA inflamatoria periférica; son
ineficaces para la enfermedad de la columna vertebral. Por el contrario, los
antagonistas del fármaco TNF-α han sido notablemente eficaces para la
enfermedad espinal en el tratamiento de las manifestaciones espinales de las
SPA. Los agentes biológicos más nuevos que se han encontrado efectivos en el
tratamiento de SPA incluyen ustekinumab, que bloquea los receptores IL-12 e
IL-23, y secukinumab e ixekizumab, anticuerpos monoclonales dirigidos contra IL-17A.
Apremilast es un inhibidor oral de molécula pequeña de la fosfodiesterasa 4, y
ha demostrado ser eficaz en el tratamiento de la psoriasis y la artritis
psoriásica.
LUPUS ERITEMATOSO SISTÉMICO
El lupus eritematoso sistémico (LES) es una enfermedad
autoinmune multisistémica caracterizada principalmente por síntomas
constitucionales, anomalías hematológicas y depósito de inmunocomplejos en
órganos diana. Suele afectar a mujeres en edad fértil. Algunos pacientes con
una deficiencia hereditaria del complemento tienen un mayor riesgo de
desarrollar LES. Las manifestaciones potenciales de SLE son innumerables (tabla
30.1), y el diagnóstico de SLE requiere una fuerte sospecha clínica, pruebas de
laboratorio o patológicas de la enfermedad y la exclusión de otras posibles
afecciones infecciosas, reumáticas y neoplásicas. Incluso en pacientes con LES
establecido, los nuevos síntomas clínicos no deben atribuirse únicamente al
LES.


Tabla 30.1. Manifestaciones del Lupus Eritematoso
Sistémico.
Las reacciones a los medicamentos y las infecciones
oportunistas ocurren con frecuencia, y el dolor articular no inflamatorio puede
indicar fibromialgia superpuesta o necrosis avascular (NOA) (Cuadro 30.4).

Cuadro 30.4. Factores de riesgo de necrosis
avascular en adultos.
La nefritis lúpica es la principal manifestación renal
del LES y se caracteriza por proteinuria y hematuria con cilindros de glóbulos
rojos. La gravedad de la enfermedad se clasifica según la evaluación
histológica de la biopsia renal para la clasificación de la nefritis por LES de
la Organización Mundial de la Salud. (cuadro 25.1)

Cuadro 25.1 Clasificación de enfermedades renales de
la Organización Mundial de la Salud
El grado de afectación renal patológica no se puede predecir de forma adecuada solo con criterios clínicos. A pesar del tratamiento, algunos pacientes todavía progresan a enfermedad renal en etapa terminal (ESRD) y requieren terapia de diálisis o trasplante renal. La trombosis de la vena renal, la nefritis intersticial y el síndrome antifosfolípido (SAF) también pueden provocar disfunción renal en pacientes con LES.
La formación de autoanticuerpos es la firma
inmunológica distintiva del LES. ANA es detectable en esencialmente el 100% de
los pacientes con LES, pero esta prueba carece de especificidad. Muchas otras
afecciones inflamatorias y más del 20 % de los pacientes sanos pueden tener un
ANA positivo. Las pruebas de anticuerpos anti-ADN de doble cadena (dsDNA) y
anti-Smith son ensayos mucho más específicos pero menos sensibles. Los anticuerpos
anti-Ro (anti-SSA), anti-La (anti-SSB) y antiproteína ribonuclear (anti-RNP)
también pueden detectarse en pacientes con LES. Una opinión prevaleciente es
que los autoanticuerpos en el lupus forman inmunocomplejos circulantes que se
depositan en los tejidos diana, reclutan complemento y otros mediadores
inmunológicos y causan enfermedad de órganos diana.
Los corticosteroides siguen siendo el pilar del
tratamiento en los brotes agudos de LES. Las dosis altas (hasta 1 mg/kg/día de
prednisona o su equivalente) a menudo se usan para anomalías hematológicas
graves o enfermedades que amenazan órganos. Las dosis más bajas de
corticosteroides o NSAID a menudo son suficientes para las manifestaciones
cutáneas, musculoesqueléticas y de serositis. Los agentes ahorradores de
esteroides se utilizan para minimizar las complicaciones relacionadas con los
esteroides. Aparte de los corticosteroides, solo la hidroxicloroquina y el
belimumab, un anticuerpo monoclonal supresor de células B anti-factor activador
de células B (BAFF), han sido aprobados para su uso en LES por la
Administración de Drogas y Alimentos de los EE. UU. (FDA). Independientemente,
muchos otros medicamentos inmunosupresores y citotóxicos se han usado con éxito
en el tratamiento del LES: estos medicamentos incluyen hidroxicloroquina,
azatioprina, micofenolato mofetil, metotrexato, ciclosporina, ciclofosfamida y
posiblemente rituximab, aunque este agente no está aprobado por la FDA para el
LES. Los agentes antipalúdicos (p. ej., hidroxicloroquina) son fármacos bien
tolerados que desempeñan un papel importante en el tratamiento del LES a través
de mecanismos poco conocidos. El metotrexato es particularmente útil en el
tratamiento de las manifestaciones cutáneas, musculoesqueléticas y de serositis
del LES. Belimumab es efectivo principalmente para las características cutáneas
y musculoesqueléticas del LES. La azatioprina, el micofenolato de mofetilo, la
ciclosporina y la ciclofosfamida tienen eficacia comprobada y suelen ser
eficaces en el tratamiento de la nefritis lúpica. La ciclofosfamida es un
fármaco citotóxico reservado para las manifestaciones más graves del LES (p.
ej., nefritis grave, hemorragia pulmonar, enfermedad grave del sistema nervioso
central [SNC], vasculitis). Las complicaciones agudas de la ciclofosfamida
incluyen cistitis hemorrágica, supresión de la médula ósea, infertilidad e
inmunosupresión profunda. El linfoma y los cánceres del tracto urinario son
eventos adversos potenciales a largo plazo. La plasmaféresis y la
inmunoglobulina intravenosa (IGIV) pueden agregarse a la terapia
inmunosupresora para el LES potencialmente mortal. La anticoagulación se
utiliza en pacientes con complicaciones trombóticas.
SAF es un estado hipercoagulable en el que un
paciente sufre un coágulo o pérdida fetal en el contexto de tener anticuerpos
antifosfolípidos séricos detectables medidos en dos pruebas separadas con al
menos 12 semanas de diferencia. Los coágulos pueden ser de naturaleza venosa,
arterial o incluso microvascular. La pérdida fetal incluye tres o más abortos
espontáneos en el primer trimestre o cualquier pérdida fetal después del primer
trimestre. Los anticuerpos antifosfolípidos incluyen anticuerpos
anticardiolipina (IgG o IgM), anticuerpos anti-beta-2-glucoproteína I o el
anticoagulante lúpico, que eleva el tiempo de tromboplastina parcial (PTT) o el
tiempo de veneno de víbora de Russell diluido y no se corrige al mezclarlo con
suero normal. SAF secundario describe una enfermedad que ocurre en presencia
de una enfermedad autoinmune subyacente, más comúnmente LES, mientras que tal
trastorno no se detecta en SAF primario. El SAF catastrófico es el SAF con múltiples coágulos, trombosis microvascular e insuficiencia
orgánica. Tiene una tasa de mortalidad reportada como 50% o mayor. Todas las
formas o manifestaciones de SAF se tratan con anticoagulación. También se
puede utilizar la inmunosupresión y añadir plasmaféresis en casos de SAF refractario o SAF catastrófico.
Finalmente, el lupus inducido por fármacos (DIL) puede
ocurrir con el uso de hidralazina, procainamida, isoniazida, metildopa,
quinidina, minociclina o incluso agentes anti-TNF-α. Se caracteriza
principalmente por síntomas constitucionales, hallazgos mucocutáneos, serositis
y elevación de ANA. La enfermedad que amenaza los órganos debe hacer que se
considere un diagnóstico alternativo. Los anticuerpos antihistona son un
marcador sensible, pero no específico, de DIL. Los síntomas generalmente se
resuelven después de la eliminación del fármaco causante.
ESCLERODERMA
La esclerodermia describe una familia de trastornos
raros pero relacionados que comúnmente comparten fibrosis dérmica idiopática.
La esclerodermia se clasifica en enfermedad localizada (varios tipos de morfea
y esclerodermia lineal) y esclerosis sistémica (SSC). La SSC es una enfermedad
caracterizada tanto por fibrosis como por vasculopatía. La SSC se subdivide en
enfermedad limitada o difusa según la extensión de la fibrosis. En SSC
limitada, la fibrosis dérmica se restringe a las manos, los pies y la cara. La
afectación de las extremidades proximales, por lo general proximal a las
articulaciones metacarpofalángicas o al tronco, indica SSC difusa. La fibrosis
de órganos representa una fuente importante de mortalidad. Ocurre
principalmente en los pulmones, el corazón y el tracto gastrointestinal (GI).
La vasculopatía explica la hipertensión pulmonar, la crisis renal
esclerodérmica y el fenómeno de Raynaud casi universal. Los pacientes con SSC
pueden manifestar los síntomas CREST (calcinosis cutis, Raynaud, dismotilidad
esofágica, esclerodactilia y telangiectasia), que tienen un alto riesgo de
desarrollar hipertensión pulmonar y también pueden ocurrir independientemente
del SSC. Los análisis serológicos de pacientes con SSC a menudo tienen ANA
positivos y autoanticuerpos más específicos: anticuerpos anticentrómero,
anti-SCL70 y anti-ARN polimerasa III. Los anticuerpos anticentrómero se asocian
más a menudo con SSC limitada, CREST e hipertensión pulmonar. Los anticuerpos
anti-SCL70 se asocian con SSC difusa y fibrosis cardiopulmonar. Los anticuerpos
anti-ARN polimerasa III se asocian con SSC cutánea rápidamente progresiva y
crisis renal.
Existen opciones de tratamiento para las
complicaciones vasculares del SSC. La crisis renal por esclerodermia se
presenta como hipertensión, hematuria e insuficiencia renal. A pesar de la
insuficiencia renal, la crisis renal se trata con inhibidores de la enzima
convertidora de angiotensina (ECA), bloqueadores de los receptores de
angiotensina o ambos. El uso intensivo de estos medicamentos para reducir la
presión arterial ha cambiado la historia natural de la crisis renal por
esclerodermia, de modo que los pacientes pueden recuperarse o incluso evitar la
terapia de diálisis.
La hipertensión pulmonar y la enfermedad de Raynaud
que amenaza los dedos se tratan con terapias vasodilatadoras. Los bloqueadores
de los canales de calcio dihidropiridínicos, los antagonistas de los receptores
alfa y la toxina botulínica inyectada pueden ser eficaces para el fenómeno de
Raynaud. Los antagonistas de los receptores de endotelina (p. ej., bosentán),
los inhibidores de la fosfodiesterasa (p. ej., sildenafil) y las prostaciclinas
también son eficaces para el fenómeno de Raynaud y han reducido notablemente la
mortalidad por hipertensión pulmonar.
Falta un tratamiento farmacológico de las
complicaciones fibróticas. Los agentes inmunosupresores, incluida la
ciclofosfamida, se han utilizado para la fibrosis pulmonar, con solo una mejora
modesta en el resultado. La terapia adicional está dirigida a los síntomas. Los
inhibidores de la bomba de protones en dosis altas se usan para la enfermedad
por reflujo gastroesofágico y deben administrarse a todos los pacientes con SSC
para disminuir el riesgo de estenosis esofágica. Los agentes promotores de la
motilidad (p. ej., metoclopramida, eritromicina) se usan para la dismotilidad
gastrointestinal y los antibióticos orales se usan para el síndrome de
sobrecrecimiento intestinal.
Varios otros síndromes pueden causar fibrosis cutánea
o sistémica y deben ser considerados en el diagnóstico diferencial de SSC. La
fascitis eosinofílica causa fibrosis cutánea y resulta de la infiltración de
eosinófilos en la fascia subcutánea. La enfermedad de injerto contra huésped causa
fibrosis cutánea e intestinal. La fibrosis inducida por gadolinio (fibrosis
sistémica nefrogénica) ocurre en algunos pacientes con insuficiencia renal
grave que están expuestos a agentes de contraste que contienen gadolinio que se
usan a menudo en procedimientos de resonancia magnética.
OTRAS ENFERMEDADES DEL TEJIDO CONECTIVO
SÍNDROME DE SJÖGREN
El síndrome de Sjögren (SS) puede ser una entidad
primaria o puede existir de forma secundaria a otras enfermedades del tejido
conectivo (ETC) como LES, AR y SSC. Síndrome Sicca describe las características más
predominantes de SS; la boca seca y los ojos secos resultan de la destrucción
autoinmune de las glándulas salivales y lagrimales. Los pacientes con SS a
menudo tienen anticuerpos ANA, anti-Ro y anti-La positivos, un FR elevado e
hipergammaglobulinemia.
Las complicaciones más graves incluyen vasculitis de
vasos pequeños, poliartritis, neuropatías periféricas, ILD y linfoma. Los
linfomas típicamente ocurren en el tejido linfoide asociado a la mucosa y
algunas veces son anunciados por una gammapatía monoclonal y una caída en el
título de FR. El tratamiento para el SS generalmente se dirige a los síntomas
con saliva artificial y lágrimas.
La vasculitis o ILD requiere altas dosis de
corticosteroides u otras terapias inmunosupresoras intensivas como la
ciclofosfamida, lo que puede aumentar el riesgo a largo plazo de linfoma en
estos pacientes.
MIOPATÍAS INFLAMATORIAS IDIOPÁTICAS
Las miopatías inflamatorias idiopáticas (MII) incluyen
una colección de enfermedades autoinmunes: dermatomiositis (DM), polimiositis,
miositis asociada a malignidad, DM juvenil y miositis por cuerpos de inclusión
(IBM). Con la exclusión de IBM, estas condiciones se presentan como debilidad
muscular proximal. La creatina quinasa (CK) suele estar elevada hasta al menos
5 a 10 veces el límite superior normal, y la aldolasa también puede estar
elevada. La DM, la miositis asociada a neoplasias malignas y la DM juvenil
pueden tener manifestaciones cutáneas. Éstos incluyen un exantema malar que, a
diferencia del LES, afecta los pliegues nasolabiales, un exantema heliotropo
violáceo periorbitario, un signo de chal fotosensible sobre el precordio, un
signo de funda eritematoso en la parte lateral del muslo, pápulas de Gottron
sobre los nudillos dorsales y manos de mecánico hiperqueratósicas.
Un subgrupo de pacientes con DM solo tiene afectación
de la piel, lo que se denomina dermatomiositis amiopática. La ILD puede ocurrir
en muchos de los diversos MII, pero por lo general ocurre en pacientes con el
anticuerpo anti-Jo-1. La presencia de otros autoanticuerpos se ha asociado con
otras manifestaciones de MII. La afectación orofaríngea por cualquiera de las MII puede poner en peligro la vida debido al riesgo de aspiración. El
diagnóstico se ayuda con resonancia magnética o electromiografía y se confirma
con una biopsia muscular que muestra un infiltrado inflamatorio.
El tratamiento consiste principalmente en
corticosteroides en dosis altas, con agentes inmunosupresores adicionales (como
metotrexato, azatioprina, IVIG o posiblemente rituximab) que se usan en
pacientes que no pueden reducir gradualmente los corticosteroides. A diferencia
de los otras MII, IBM se presenta como debilidad muscular distal y atrofia en
pacientes de edad avanzada. El diagnóstico se realiza mediante microscopía
electrónica de una biopsia muscular. El tratamiento es generalmente ineficaz.
ENFERMEDAD DE STILL DE INICIO EN ADULTOS
La enfermedad de Still del adulto (ESA) es una
enfermedad inflamatoria multisistémica sistémica caracterizada por fiebre alta,
dolor de garganta previo, erupción cutánea evanescente, linfadenopatía,
hepatoesplenomegalia, artritis inflamatoria, transaminasas hepáticas elevadas,
leucocitosis neutrofílica y marcadores de laboratorio marcadamente anormales de
inflamación, especialmente ferritina. (ver cuadro 30.1). Es un diagnóstico de
exclusión tras descartar AR, LES y enfermedades infecciosas y malignas.
El tratamiento es similar al de la AR, pero puede
requerir dosis más altas de esteroides. En particular, la enfermedad de Still del adulto puede responder
de manera bastante espectacular al antagonista del receptor de IL-1 anakinra o
al anticuerpo contra el receptor de IL-6 tocilizumab.
ENFERMEDAD MIXTA DEL TEJIDO CONECTIVO
La enfermedad mixta del tejido conectivo (EMTC) es un
ejemplo de un síndrome de superposición que se manifiesta con varios elementos
de varias enfermedades autoinmunes. Las características pueden incluir artritis
inflamatoria, esclerodactilia, síndrome de Raynaud, miositis inflamatoria,
hipertensión pulmonar y síndrome de Sjögren secundario. La serología es notable
por un título muy alto de ANA positivo y un anticuerpo anti-RNP de título alto,
pero estos hallazgos aún carecen de cierta especificidad. El tratamiento está
dirigido a las manifestaciones individuales de la EMTC.
VASCULITIS
Colectivamente, las vasculitis representan una
colección de enfermedades caracterizadas por la inflamación de los vasos
sanguíneos. Las vasculitis a menudo se clasifican en función de su afectación
de vasos grandes, medianos o pequeños (tabla 30.2).

Tabla 30.2. Categorización de las Vasculitis
Sistémicas
La demografía del paciente y el análisis serológico
son características distintivas adicionales. La mayoría de las vasculitis son
afecciones graves que ponen en peligro los órganos o la vida. Las fiebres, los
síntomas constitucionales y los marcadores inflamatorios anormales (v. cuadro
30.1) son características comunes de la mayoría de las vasculitis. Un
diagnóstico definitivo a menudo requiere evidencia de inflamación vascular por
biopsia. La inmunosupresión con corticoides es un pilar del tratamiento. A
menudo se agregan agentes ahorradores de esteroides (azatioprina, metotrexato,
micofenolato mofetilo, rituximab) o agentes citotóxicos (ciclofosfamida) para
limitar la toxicidad de los esteroides y proporcionar inmunosupresión
adicional. En su mayor parte, los bloqueadores de TNF-α son ineficaces en la
vasculitis sistémica.
VASCULITIS DE GRANDES VASOS
ARTERITIS DE CÉLULAS GIGANTES
La arteritis de células gigantes (ACG) es la
vasculitis sistémica más frecuente. Los pacientes afectados son exclusivamente
mayores de 50 años y por lo general tienen ascendencia del norte de Europa. Los
pacientes suelen caer en uno de los tres tipos de presentaciones: arteritis
craneal, aortitis o fiebre de origen desconocido (FOD). Aproximadamente el 30 %
de los pacientes con ACG tendrán PMR concurrente o preexistente, y entre el 15
% y el 20 % de los pacientes con PMR desarrollarán ACG. Por lo tanto, los
pacientes con PMR deben ser interrogados de forma rutinaria acerca de los
síntomas de la arteritis craneal.
Los síntomas craneales o cambios visuales en un
paciente con PMR se consideran una emergencia reumatológica. Los síntomas de la
arteritis craneal incluyen sensibilidad en el cuero cabelludo, dolor de cabeza
de nueva aparición, claudicación mandibular, tos persistente y cambios
visuales.
Una complicación temida es la afectación de la arteria
oftálmica, que puede provocar ceguera irreversible. La ACG puede presentarse
como claudicación del brazo o tos, pero la aortitis en sí misma puede ser
asintomática y encontrarse en patología posquirúrgica. En pacientes >65
años, la ACG comprende del 15% al 20% de los casos de FOD.
Los marcadores inflamatorios suelen estar bastante
elevados en la ACG (ver Tabla 30.1). Independientemente de la presentación, el
diagnóstica de ACG se confirma mediante la evaluación histológica de la
vasculatura afectada. La biopsia unilateral o, a veces, bilateral de la arteria
temporal a menudo confirmará el diagnóstico en pacientes con síntomas craneales
o FOD. La modalidad primaria de tratamiento son los corticosteroides
(prednisona, 1 mg/kg/d) reducidos gradualmente durante varios meses.
Tocilizumab, el anticuerpo anti-receptor de IL-6, ha demostrado recientemente
ser un tratamiento eficaz para la ACG. El metotrexato y la azatioprina pueden
considerarse agentes ahorradores de esteroides para pacientes que no pueden
reducir los esteroides de manera razonable. Los antagonistas de TNF-α son
ineficaces y aumentan la tasa de infecciones graves.
ARTERITIS DE TAKAYASU
La arteritis de Takayasu es una vasculitis de grandes
vasos que suele afectar a mujeres jóvenes, lo que la distingue de la ACG. La
presentación clásica es la de claudicación de las extremidades en un paciente
con presión arterial asimétrica, soplos vasculares o falta de pulso. Las
arterias mesentéricas proximales o renales también pueden estar involucradas.
La inflamación arterial crónica conduce a estenosis fibróticas, que explican
los síntomas. El tejido puede ser difícil de obtener y el diagnóstico puede
basarse en estudios de imágenes (angiografía convencional, angiografía por TC,
angiografía por RM y tomografía por emisión de positrones [PET] TC). La VSG y
la proteína C reactiva (PCR) pueden estar elevadas, pero no de manera tan
confiable como en la ACG. Los corticosteroides también son la principal
modalidad de tratamiento, aunque varios medicamentos biológicos también pueden
considerarse agentes ahorradores de esteroides.
VASCULITIS DE VASOS MEDIANOS
POLIARTERITIS NODOSA
La poliarteritis nodosa (PAN) es la vasculitis
prototípica de vasos medianos. Un subgrupo de pacientes con PAN tiene infección
documentada por el virus de la hepatitis B. Los pacientes con PAN suelen
presentar síntomas constitucionales, lesiones cutáneas purpúricas e
insuficiencia renal. La afectación de órganos adicionales puede incluir la
vasculatura pulmonar, el intestino, la vesícula biliar, los testículos o los
ovarios. Existe una variante de PAN limitada a la piel que se localizasólo en la
piel (PAN cutánea). El estudio serológico de vasculitis no es destacable. El
diagnóstico puede estar respaldado por un angiograma renal o mesentérico que
muestre enfermedad aneurismática. La biopsia del tejido afectado muestra
vasculitis necrotizante de vasos de tamaño mediano. El tratamiento consiste en
corticosteroides, con agentes ahorradores de esteroides o citotóxicos agregados
para casos más graves o refractarios.
ENFERMEDAD DE KAWASAKI
La enfermedad de Kawasaki es una vasculitis de la
infancia, aunque se han notificado casos en adolescentes y adultos jóvenes. Es
una vasculitis de vasos medianos que se presenta como fiebre >5 días,
conjuntivitis, exantema descamativo en las extremidades, edema periférico y
lengua fresa eritematosa en el contexto de VSG y PCR elevadas. La afectación
de las arterias coronarias puede causar aneurismas o estenosis potencialmente
mortales. Aunque los corticosteroides son útiles para controlar los síntomas,
el uso combinado de aspirina e IVIG ha cambiado la historia natural de esta
enfermedad al reducir notablemente la incidencia de complicaciones de las
arterias coronarias.
VASCULITIS DE PEQUEÑOS VASOS
Las vasculitis que afectan las arteriolas, los
capilares y las vénulas se clasifican en dos categorías de enfermedades: las
asociadas con anticuerpos anticitoplasma de neutrófilos (ANCA) y las asociadas
con el depósito de inmunocomplejos. Es particularmente importante excluir la
endocarditis infecciosa en el estudio de una vasculitis de vasos pequeños.
VASCULITIS ASOCIADAS A ANCA
Las vasculitis asociadas a ANCA (VAA) incluye la enfermedad granulomatosa con poliangeítis (GPA, anteriormente granulomatosis de Wegener
[WG]), poliangeítis microscópica (MPA) y la granulomatosis eosinofílica con
poliangeítis (anteriormente síndrome de Churg-Strauss [CSS]), VAA inducida por
fármacos, y VAA específicos de órganos (como VAA renal aislada). Las VAA causan
vasculitis necrotizante de los tejidos diana, que pueden incluir los senos
paranasales, la órbita, las vías respiratorias superiores, los alvéolos, el
miocardio, los glomérulos, el SNC, los nervios periféricos, el tubo digestivo y
la piel. Las causas de afectación renal pueden progresar a una
glomerulonefritis pauciinmune semilunar aguda y pueden conducir a una ESRD. Las
complicaciones potencialmente mortales incluyen hemorragia pulmonar,
miocarditis, vasculitis mesentérica y vasculitis del SNC. Las lesiones pueden
ser de naturaleza granulomatosa o, en el caso de CSS, principalmente
eosinofílicas. Los pacientes pueden tener artralgias asociadas y evidencia de laboratorio
de inflamación sistémica (ver Cuadro 30.1).
El estudio serológico para VAA a menudo revela la
presencia de ANCA en un patrón de ANCA citoplásmico (cANCA), ANCA perinuclear
(pANCA) o ANCA no específico. Los ANCA pueden asociarse con anticuerpos antiproteinasa-3
(anti-PR3) o antimieloperoxidasa (anti-MPO) mediante ensayo inmunoabsorbente
ligado a enzimas (ELISA). Un patrón cANCA con ELISA anti-PR3 es altamente
específico para WG/GPA, mientras que las otras AAV tienden a mostrar un patrón
pANCA con ELISA anti-MPO. Se pueden observar patrones de ANCA o pANCA no
específicos en ausencia de especificidad ELISA en otras afecciones
inflamatorias, como colitis ulcerosa, colangitis esclerosante primaria y
vasculitis inducida por fármacos por agentes como la minociclina, el alopurinol
o el propiltiouracilo. El levamisol, que a menudo se usa como contaminante de
la cocaína, puede causar una VAA inducida por fármacos, a menudo asociada con
anticuerpos duales anti-PR3 y anti-MPO. Por lo general, se presenta como lesiones
cutáneas, pero puede progresar a glomerulonefritis y hemorragia pulmonar. Los
anticuerpos antimembrana basal glomerular también deben testearse en pacientes
con VAA y síntomas pulmonares y renales. Los niveles de complemento son
normales o elevados en VAA.
La terapia inmunosupresora es fundamental para el
tratamiento de las VAA. Las dosis altas de corticosteroides (prednisona, 1
mg/kg/día) y la ciclofosfamida o el rituximab se usan para inducir la remisión
en cualquier paciente con enfermedad que amenaza la vida o los órganos. Se
puede agregar plasmaféresis a aquellos que son refractarios al tratamiento. Una
vez que se induce la remisión, la ciclofosfamida puede sustituirse por
rituximab, metotrexato o azatioprina para minimizar la toxicidad a largo plazo
de la ciclofosfamida. El metotrexato tiene un papel en el tratamiento de la
enfermedad VAA más limitada.
VASCULITIS DE PEQUEÑOS VASOS MEDIADAS POR COMPLEJOS
INMUNE
Las vasculitis de vasos pequeños mediadas por
inmunocomplejos pueden ser causadas por varios procesos patológicos diferentes.
Estos incluyen púrpura de Henoch-Schönlein (HSP), vasculitis crioglobulinémica,
vasculitis por hipersensibilidad y vasculitis asociada con enfermedades del
tejido conectivo (p. ej., LES, AR, síndrome de Sjögren). Estas condiciones
tienen en común el depósito de inmunocomplejos en los tejidos diana. Los
niveles de complemento sérico a menudo están disminuidos y se correlacionan con
la actividad de la enfermedad. La HSP es una vasculitis de vasos pequeños
mediada por IgA que afecta principalmente a la piel, los riñones y el tubo
digestivo en forma de púrpura, glomerulonefritis y diarrea sanguinolenta. Por
lo general, es un trastorno autolimitado, pero los síntomas graves pueden
requerir una terapia con corticosteroides.
La crioglobulinemia puede causar una vasculitis
caracterizada por glomerulonefritis, neuritis periférica, púrpura y rara vez
vasculitis mesentérica. Las crioglobulinas de tipo II (mixtas) asociadas al
virus de la hepatitis C (VHC) son las que tienen más probabilidades de causar
vasculitis.
El tratamiento está dirigido a reducir la carga viral
del VHC. La vasculitis por hipersensibilidad es típicamente inducida por
fármacos y limitada a la piel como la denominada vasculitis leucocitoclástica.
La eliminación del agente agresor es curativa. Las vasculitis asociadas a ETC se analizan por separado en cada tema de enfermedad.
OTRAS ARTRITIS INFLAMATORIAS
ARTRITIS MICROCRISTALINA
La gota y la seudogota típicamente se presentan como
una monoartritis inflamatoria exuberante, aunque los pacientes pueden tener
enfermedad poliarticular. La artritis gotosa ocurre debido a una reacción
inflamatoria contra los cristales de urato monosódico. La seudogota tiene una
presentación similar, pero los cristales patógenos son dihidrato de pirofosfato
de calcio (CPPD). En ambos casos, la inflamación sistémica por otras causas (es
decir, infección concurrente) puede precipitar un ataque de artritis
microcristalina. La artritis gotosa tiende a afectar las extremidades inferiores,
mientras que la pseudogota prefiere las rodillas, las muñecas y las manos.
Aunque cualquier articulación puede verse afectada por la gota o la seudogota,
los hombros, las caderas y la columna suelen estar libres de gota.
El diagnóstico de artritis microcristalina se confirma
mediante aspirado de líquido sinovial y observación de cristales mediante
microscopía de luz polarizada compensada. En el caso de la gota, los cristales
son largos, con forma de aguja, brillantes y con birrefringencia negativa.
Habitualmente, esto significa que los cristales son amarillos cuando están en
el plano de la luz polarizante y azules cuando son perpendiculares. Por otro
lado, los cristales de la enfermedad de CPPD son cortos, romboides, tenues y
positivamente birrefringentes (amarillos cuando están perpendiculares al
polarizador). Tanto en la gota como en la seudogota, el líquido articular es
inflamatorio, a menudo con 10 a 50 × 103 WBC/mm3 de líquido. La artritis
séptica concurrente debe excluirse mediante tinción de Gram y cultivo de
líquido, en especial si el WBC líquido es >100 × 103 WBC/mm3.
El nivel de ácido úrico en suero tiene poco papel en
el diagnóstico de la artritis gotosa aguda porque los niveles de ácido úrico
durante un ataque pueden no representar los niveles de referencia. La
condrocalcinosis puede estar presente en una radiografía simple de un paciente
con seudogota, pero también puede encontrarse en pacientes asintomáticos.
La artritis aguda por gota o seudogota puede tratarse
con AINE, corticosteroides o colchicina oral. Los AINE son efectivos y seguros
en pacientes sin coagulopatía, insuficiencia renal o úlcera péptica. Los
corticosteroides pueden administrarse sistémicamente en dosis bajas (prednisona
20 mg por día o menos) o por inyección intraarticular. Ambos son efectivos,
aunque el primero es más apropiado para ataques poliarticulares. La colchicina
oral es menos eficaz que las otras y juega un papel más importante en la
profilaxis contra los ataques de gota o en la interrupción de un ataque
inminente si se toma al primer signo de síntomas. Ya no es apropiado “dosificar
a la diarrea”. Debe ajustarse la dosis para la insuficiencia renal y puede
causar supresión de la médula ósea, toxicidad hepática o incluso la muerte si
se toma en cantidades excesivas. La colchicina intravenosa nunca debe usarse
debido a su perfil de toxicidad, incluidas las arritmias cardíacas. La
hiperuricemia es un factor de riesgo para la gota. El ácido úrico sérico puede
reducirse mediante terapias antihiperuricémicas que incluyen alopurinol,
febuxostat o probenecid, lesinurad y pegloticasa. Estas terapias
antihiperuricémicas (alopurinol, febuxostat, lesinurad, probenecid y
pegloticasa) generalmente se reservan para personas con artritis gotosa
erosiva, nefropatía por ácido úrico, nefrolitiasis por ácido úrico, gota
tofácea o ataques frecuentes de gota. No deben iniciarse ni ajustarse durante
un episodio de artritis gotosa. Más bien, se agregan varias semanas después del
ataque, con las medidas profilácticas apropiadas de colchicina, AINE o
esteroides en dosis bajas. El alopurinol y el febuxostat, ambos inhibidores de
la xantina oxidasa, se excretan por vía renal. Ambos agentes pueden causar
supresión de la médula ósea y hepatotoxicidad, mientras que el alopurinol se
asocia con una reacción de hipersensibilidad grave que puede poner en peligro
la vida si no se identifica. Ambos agentes potencian el efecto de la
azatioprina o la mercaptopurina, por lo que estas combinaciones deben usarse
con mucha precaución. El probenecid y el lesinurad son fármacos uricosúricos
que por lo general son ineficaces en pacientes con algún grado de insuficiencia
renal moderada (creatinina >2,0 mg/dl). La pegloticasa es una enzima uricasa
recombinante acoplada a polietilenglicol.
Aunque es muy eficaz para reducir el ácido úrico, se
asocia con un riesgo muy alto de anafilaxia durante la infusión del fármaco.
Con cualquier agente antihiperuricémico, el ácido úrico sérico objetivo es al
menos <6 mg/dl, o <5 mg/dl si hay tofos.
ARTRITIS INFECCIOSA
La artritis infecciosa debe encabezar la lista de
preocupaciones en cualquier paciente con monoartritis aguda. La artritis
séptica bacteriana es una emergencia reumatológica. La artrocentesis es
obligatoria si se sospecha artritis séptica. Los factores de riesgo son
similares a los de la endocarditis, incluido el uso de drogas por vía
intravenosa, la inmunosupresión, la ruptura de las barreras mucocutáneas y la
actividad sexual de alto riesgo. Las personas con prótesis articulares y las
afectadas por otros procesos inflamatorios (p. ej., AR) también están en
riesgo. Aunque cualquier articulación puede estar involucrada, las
articulaciones grandes se ven más comúnmente afectadas que las articulaciones
pequeñas. Los patógenos bacterianos comunes incluyen Staphylococcus aureus,
especies de Streptococcus y especies de Neisseria. En estos casos, los WBC
líquidos sinoviales suelen ser muy altos (>50 × 103 WBC/mm3) con >95% de
leucocitos polimorfonucleares. La tinción de Gram del líquido sinovial puede mostrar
microorganismos y el cultivo del líquido es diagnóstico. Las especies de
Neisseria requieren agar chocolate para su crecimiento. Los cultivos pueden ser
negativos en pacientes que han recibido antibióticos como antecedente. Es
prudente buscar una fuente bacteriana porque la artritis séptica suele ser el
resultado de la siembra hematógena de una articulación. La artritis séptica se
trata con aspiración articular, antibióticos intravenosos y, a menudo,
artrotomía quirúrgica. La infección de una articulación protésica es
particularmente problemática y generalmente justifica la extracción de la prótesis y antibióticos prolongados.
Las espiroquetas, las micobacterias, los hongos y los
parásitos pueden causar una artritis séptica más crónica. Por ejemplo, la
presentación típica de la artritis de Lyme (de Borrelia burgdorferi) es la de
una monoartritis crónica con hinchazón fuera de proporción con el dolor. La
artritis de Lyme tiende a afectar la rodilla u otras articulaciones grandes. Es
una manifestación de la enfermedad de Lyme crónica que ocurre meses después de
la picadura de la garrapata. El diagnóstico se basa en la presentación clínica
y la presencia de serología Lyme positiva por ELISA y Western blot. Un mes de
doxiciclina oral es el tratamiento de primera línea para la artritis de Lyme.
La ceftriaxona intravenosa puede usarse en casos refractarios al tratamiento.
También se puede desarrollar una artritis de Lyme posinfecciosa y se trata con drogas modificadoras de la enfermedad como la hidroxicloroquina o el metotrexato, o incluso con terapias
biológicas. En casos de monoartritis crónica no diagnosticada, la biopsia de
tejido sinovial está indicada para evaluar la presencia de micobacterias,
hongos y parásitos.
La poliartritis es una presentación menos común de la
artritis séptica bacteriana, aunque no es inusual en el caso de la artritis
viral. El virus de la hepatitis B, el VHC, el virus de la inmunodeficiencia
humana, el parvovirus B19 y el virus de la rubéola (y la vacuna) se relacionan
con una verdadera poliartritis inflamatoria, mientras que muchos otros virus
pueden causar poliartralgias sin artritis manifiesta. Puede ocurrir
poliartritis séptica de origen bacteriano, pero es mucho menos común. Se asocia
con un mal resultado porque refleja un alto grado de bacteriemia.
TRASTORNOS REUMÁTICOS NO INFLAMATORIOS
OSTEOARTRITIS
La osteoartritis (OA) es, con mucho, la causa más
común de artritis en los Estados Unidos. Es el resultado de fuerzas mecánicas
locales anormales que causan la degeneración de las articulaciones y lesiones
del cartílago con el tiempo. Los factores de riesgo para la OA incluyen la
edad, la obesidad, los antecedentes familiares, los traumatismos repetitivos,
los trastornos articulares internos y la artritis inflamatoria previa (p. ej., AR, SPA, artritis séptica). La OA es una artritis crónica que generalmente
afecta las rodillas, las caderas, la columna vertebral, los pulgares, las
articulaciones interfalángicas proximales y las articulaciones interfalángicas
distales de las personas mayores. La OA en personas más jóvenes o la OA que
afecta las muñecas, los codos, los hombros o los tobillos es poco común. Tal
afectación puede indicar OA secundaria como resultado de un trastorno articular
interno no reconocido, artritis inflamatoria, anomalía congénita, necrosis
avascular, condrocalcinosis, hemocromatosis u ocronosis por alcaptonuria.
Las opciones de tratamiento no quirúrgico en la OA son
limitadas. La fisioterapia, la reducción de peso, los cambios en el estilo de
vida, los AINE y las inyecciones de corticosteroides pueden ser intervenciones
útiles.
Los analgésicos opiáceos deben usarse con moderación.
Los nutracéuticos como la glucosamina no tienen ningún beneficio comprobado. La
colocación de prótesis articulares puede ser necesaria en pacientes con dolor
debilitante que afecta la función.
TRASTORNOS MUSCULOESQUELÉTICOS REGIONALES
Las quejas de tejidos blandos representan un
porcentaje sustancial de las visitas al consultorio del médico. A diferencia de
la artritis inflamatoria, la rigidez matutina es de corta duración y los síntomas
tienden a exacerbarse con la actividad. El examen debe incluir la región
afectada, así como las estructuras proximales y distales.
Procesos como la tendinitis y la bursitis suelen
mejorar con hielo, analgésicos tópicos, AINE, reposo o entablillado.
La terapia física u ocupacional es un complemento
importante. Las inyecciones de corticosteroides generalmente brindan solo un
alivio temporal, incluso en casos refractarios.
Los síndromes comunes de tejidos blandos de la
extremidad superior proximal incluyen bursitis subacromial, pinzamiento del
tendón del supraespinoso, tendinitis del bíceps, desgarro del manguito de los
rotadores o patología de la columna cervical. La inyección de lidocaína en el
hombro debe permitir que el paciente supere cualquier déficit funcional si los
síntomas están relacionados con bursitis subacromial o pinzamiento del
tendón del supraespinoso, pero no con el desgarro del manguito de los
rotadores.
Los pacientes con síndromes de tejido blando del
hombro se benefician enormemente de la fisioterapia. Es crucial no entablillar
el hombro en exceso ya que corre el riesgo de capsulitis adhesiva (hombro
congelado).
Los síndromes comunes de tejidos blandos de la
extremidad superior distal incluyen epicondilitis lateral (codo de tenista),
epicondilitis medial (codo de golfista) y síndrome del túnel carpiano (STC). A
pesar de sus nombres, todas estas condiciones representan síndromes de uso
excesivo que surgen de la muñeca. La epicondilitis se exacerba con la
resistencia isométrica contra la muñeca. El STC se presenta como parestesias en
la distribución del nervio mediano y es provocado por una prueba de Phalen o
Tinel. Las férulas para la muñeca son un tratamiento de primera línea adecuado
para estos trastornos. El STC bilateral en un paciente sin antecedentes de uso
excesivo puede ser el primer signo de una tenosinovitis inflamatoria, como
puede ocurrir en la AR u otros trastornos de retención de líquidos, como el
embarazo o el hipotiroidismo.
Los síndromes comunes de tejidos blandos de la
extremidad inferior proximal incluyen la bursitis trocantérica y el síndrome de
la banda iliotibial. En la bursitis trocantérica, los pacientes se quejan de
dolor focal sobre la bursa trocantérea lateral que empeora cuando duerme de ese
lado o cuando se palpa directamente. A menudo refleja una mala mecánica de la
espalda y responde a la fisioterapia de la espalda. El síndrome de la banda
iliotibial es un síndrome de uso excesivo que afecta la parte lateral del muslo
y la rodilla en ciclistas y corredores ávidos. Los AINE, el hielo, los
estiramientos y la modificación de las actividades pueden brindar alivio.
Los síndromes comunes de tejidos blandos de la
extremidad inferior distal incluyen bursitis anserina, síndrome del túnel
tarsiano y fascitis plantar. Los pacientes con bursitis anserina experimentan
dolor en el tejido blando medial justo distal a la rodilla. Es probable que
refleje una mala mecánica del pie y puede responder favorablemente a las
ortesis. El síndrome del túnel tarsiano es análogo al STC y resulta del
pinzamiento del nervio tibial posterior. La fascitis plantar provoca dolor
plantar al despertar y con exceso de actividad. El estiramiento y el calzado de
apoyo son útiles.
FIBROMIALGIA
La fibromialgia es un trastorno no inflamatorio en el
que los pacientes experimentan dolor miofascial musculoesquelético crónico sin
una fuente musculoesquelética identificable. Los avances de la investigación
respaldan la noción de que la fibromialgia representa uno de los muchos
trastornos crónicos centralizados disfuncionales de amplificación del dolor que
pueden surgir de un bajo umbral del dolor en el SNC del cerebro. Las
condiciones asociadas incluyen lumbago crónico, síndrome del intestino
irritable, trastorno afectivo estacional, síndrome de fatiga crónica, cistitis
intersticial y dolor torácico no cardiaco. Los pacientes a menudo están
desacondicionados, tienen sobrepeso y tienen un trastorno del estado de ánimo
coexistente. El dolor y la fatiga son síntomas dominantes de la fibromialgia.
El examen físico puede evocar respuestas de dolor exageradas a una presión
menor (puntos dolorosos desencadenantes) a lo largo del tejido blando de la
parte superior del tórax, la parte baja de la espalda y las extremidades
proximales. El estudio exhaustivo con estudios de laboratorio y de imágenes no
es revelador y, a menudo, no está justificado. Los agentes inmunosupresores no
tienen ningún beneficio y deben evitarse los analgésicos opiáceos. La terapia
dirigida por neurotransmisores es beneficiosa. Las farmacoterapias están
dirigidas a la señalización de neurotransmisores, y los agentes efectivos
incluyen gabapentina, pregabalina e inhibidores de la recaptación de serotonina
y noradrenalina (p. ej., milnaciprán y duloxetina), así como antidepresivos
tricíclicos. Las modificaciones no farmacológicas del estilo de vida son un
aspecto de importancia crítica en el tratamiento de la fibromialgia y deben
recomendarse a la mayoría de los pacientes: estado físico, fisioterapia, sueño
adecuado, dieta y pérdida de peso. Algunos pacientes se benefician de la
terapia conductual cognitiva, la biorretroalimentación, la terapia de masajes,
la acupuntura o la corrección de la apnea obstructiva del sueño. Finalmente, no
se puede exagerar la importancia de tranquilizar a los pacientes con
fibromialgia, a quienes a menudo les preocupa que tengan un trastorno maligno
no diagnosticado o una afección reumática inflamatoria sistémica como
explicación de su dolor miofascial crónico.
RESUMEN
Los síntomas musculoesqueléticos son una causa común
para que los pacientes busquen atención médica. Es importante que el médico
tratante determine si las quejas de un paciente están relacionadas con un
trastorno inflamatorio o no inflamatorio. La historia clínica, el examen físico
y algunas pruebas de laboratorio simples proporcionan los mejores medios para
hacer esta distinción. Las condiciones inflamatorias se asocian con síntomas
agudos o subagudos, rigidez matutina y articulaciones enrojecidas, calientes e
hinchadas. Los marcadores inflamatorios como ESR y CRP a menudo están elevados.
La artrocentesis es un medio seguro y eficaz para determinar si un derrame articular
es inflamatorio y facilita el diagnóstico de artritis microcristalina o
séptica. La terapia inmunosupresora a menudo está justificada para afecciones
reumáticas inflamatorias no infecciosas, como la enfermedad del tejido
conectivo, la vasculitis y la enfermedad microcristalina. La inmunosupresión
generalmente es ineficaz para los trastornos no inflamatorios, que responden
más a los AINE, la fisioterapia y las modificaciones del estilo de vida.
FUENTE:
Rheumatology Summary
DERRICK J. TODD AND JONATHAN S. COBLYN
The
Brigham Intensive Review of
Internal Medicine. (2022)
Ajay K. Singh, MBBS, FRCP, MBA
Joseph Loscalzo, MD, PhD