En este ejercicio clínico se presenta un caso que es discutido por un médico internista al que se le van proporcionando datos de la historia clínica en forma secuencial, y este analiza el cuadro a la luz de los nuevos elementos, de una manera análoga al proceso diagnóstico en la práctica real de la medicina
Un hombre de 64 años se presentó en una clínica de
reumatología con una historia de 1 año de poliartralgias migratorias. Informó
que tenía dolor, hinchazón y enrojecimiento en la cadera izquierda, seguidos de
dolor e hinchazón en la cadera derecha, lo que resultó en una capacidad
limitada para deambular sin un andador. Posteriormente se desarrolló dolor en
ambas rodillas, con hinchazón y limitación del movimiento, que posteriormente
migró para afectar ambos hombros.
PONENTE
El paciente presentó poliartritis migratoria. Un
diagnóstico diferencial no exhaustivo incluye artritis reumatoide, lupus eritematoso
sistémico, endocarditis bacteriana subaguda, artritis cristálica,
espondiloartropatías y enfermedad de Lyme. La fiebre reumática es poco probable
en un adulto mayor que vive en los Estados Unidos. La duración de los síntomas
es inconsistente con artritis viral o infección gonocócica diseminada.
EVOLUCIÓN
El paciente refirió fatiga pero no exantema, disnea,
dolor torácico, disuria, fiebre, escalofríos o síntomas gastrointestinales.
Estaba casado y trabajaba en un escritorio, participaba en actividades mínimas
al aire libre y no tenía mascotas ni exposición a animales de granja. No tenía
antecedentes de tabaquismo o uso de drogas ilícitas y reportó un consumo mínimo
de alcohol. Su madre y su hermano tenían artritis reumatoide y su hijo tenía la
enfermedad de Crohn. El examen físico mostró enrojecimiento, calor e hinchazón
de ambos hombros con rangos restringidos de abducción, rotación interna y
flexión. Otras articulaciones, incluidas las caderas y las rodillas, parecían
normales. Los sonidos del corazón eran normales. No se encontraron lesiones en
la piel. Se encontró que la velocidad de sedimentación globular (VSG) y el
nivel de proteína C reactiva estaban elevados, y el nivel de albúmina era bajo.
Un conteo sanguíneo completo, un perfil químico de la sangre de rutina, y el
nivel de creatina quinasa estaban en el rango normal. Las pruebas serológicas
para factor reumatoide, anticuerpos antipéptido cíclico citrulinado,
anticuerpos antinucleares, anticuerpos anticitoplasma de neutrófilos, HLA-B27 y
hepatitis B y C fueron negativas. La radiografía mostró derrames en la
articulación glenohumeral en ambos hombros. Se inició tratamiento con 20 mg de
prednisona al día por presunta polimialgia reumática, sin mejoría sintomática.
PONENTE
La falta de respuesta a la prednisona hace que la
polimialgia reumática sea poco probable. Dados los resultados negativos de las
pruebas serológicas autoinmunes, la infección (incluida la enfermedad de Lyme,
la endocarditis bacteriana subaguda, la infección por el virus de la inmunodeficiencia
humana [VIH] o la infección por micobacterias o hongos), o una enfermedad
inflamatoria como la artritis reumatoide seronegativa es la que más
probablemente explique su presentación. La afectación de la articulación del
hombro es menos común en pacientes con artritis cristálica, y el paciente no
tiene otros hallazgos que sugieran gota o seudogota, incluidos tofos o
condrocalcinosis en las imágenes.
EVOLUCIÓN
Se inició tratamiento con metotrexato y se aumentó la
dosis de prednisona a 40 mg, por preocupación por artritis reumatoide
seronegativa. Posteriormente se suspendió el metotrexato debido a la falta de
mejoría clínica y las elevaciones persistentes de los marcadores inflamatorios,
pero se continuó con la prednisona en una dosis de 40 mg diarios. Seis meses
más tarde, mientras el paciente todavía tomaba prednisona, se desarrollaron cuadros
febriles recurrentes, escalofríos, tos seca, calambres abdominales,
estreñimiento, pérdida de peso involuntaria y sudores nocturnos. Tenía
poliartralgia persistente sin artritis. El recuento de glóbulos blancos fue de
23,9 × 10 9 por litro (95 % de neutrófilos) y el nivel de hemoglobina fue de
10,5 g por decilitro (volumen corpuscular medio, 62 fl). Su colonoscopia más
reciente, que fue normal, se había realizado 6 años antes.
PONENTE
Con la aparición de síntomas adicionales y la
leucocitosis con predominio de neutrófilos, sigo preocupado por una infección
indolente; otras posibilidades incluyen artritis, vasculitis y cáncer asociados
con la enfermedad inflamatoria intestinal. Su uso a largo plazo de prednisona
sin diagnóstico, especialmente en ausencia de un beneficio aparente, es
preocupante. Se necesita una evaluación de posibles causas infecciosas. La
enfermedad de Whipple también debe considerarse en cualquier paciente con
poliartritis migratoria seronegativa refractaria a (o exacerbada por)
inmunosupresión; su dolor abdominal y pérdida de peso también son sugestivos de
enfermedad de Whipple, aunque su estreñimiento sería atípico.
EVOLUCIÓN
Dos muestras de hemocultivos bacterianos y uno para
hongos fueron negativos, al igual que las pruebas de anticuerpos contra el VIH,
anticuerpos contra la enfermedad de Lyme y 1,3-β- d -glucano sérico; pruebas
serológicas para el virus de Epstein-Barr, histoplasma, blastomices y
cryptococcus; y pruebas de reacción en cadena de la polimerasa (PCR) para
rickettsia, bartonella y babesia. Se enviaron hemocultivos para micobacterias.
El nivel de hierro fue de 12 μg por decilitro (rango de referencia, 50 a 150 μg
por decilitro), la capacidad total de unión de hierro 224 μg por decilitro (rango
de referencia, 250 a 400 μg por decilitro), la saturación de transferrina 5 %,
el nivel de ferritina 220 μg por decilitro (rango de referencia, 24 a 336) y el
nivel de receptor de transferrina soluble 6,0 mg por litro (rango de
referencia, 1,8 a 4,6). La tomografía computarizada (TC) de tórax, abdomen y
pelvis mostró engrosamiento circunferencial de la pared del esófago distal,
varios ganglios linfáticos mesentéricos centrales levemente prominentes,
bronquiolitis leve generalizada y un pequeño derrame pleural en el pulmón
izquierdo. La endoscopia superior mostró mucosa de aspecto normal en el
esófago, el estómago y el duodeno. Se obtuvo un número no especificado de
muestras de biopsia duodenal que mostraron duodenitis; no se observaron
macrófagos positivos para ácido peryódico de Schiff (PAS). PCR paraTropheryma
whipplei no se realizó. Una nueva colonoscopia fue normal. La tomografía por
emisión de positrones con 18 F-fluorodesoxiglucosa (FDG) con TC (FDG-PET-CT)
mostró un aumento de la captación en los ganglios linfáticos mesentéricos.
PONENTE
El nivel normal de ferritina y la baja capacidad total
de unión al hierro son consistentes con la anemia de la inflamación crónica,
pero la baja saturación de transferrina, los niveles elevados del receptor de
transferrina soluble y el volumen corpuscular medio muy bajo sugieren una
deficiencia de hierro concomitante. En este punto, se han descartado la
enfermedad inflamatoria intestinal y las causas infecciosas comunes. La
ausencia de tinción PAS en muestras de biopsia de intestino delgado no descarta
la enfermedad de Whipple; las pruebas negativas falsas son comunes,
especialmente en la enfermedad que es predominantemente extraintestinal; Se
necesitan pruebas de PCR para evaluar más a fondo esta condición. Los
hemocultivos negativos hacen que la endocarditis sea menos probable, aunque la
endocarditis infecciosa con hemocultivos negativos causada por bacterias
fastidiosas que requieren medios especializados (p. ej., Coxiella burnetii
,Mycoplasma pneumoniae , especies de brucella y Legionella pneumophila ) sigue
siendo posible. El tratamiento con prednisona debe reducirse y suspenderse,
dada la falta de beneficio y los riesgos asociados, incluida la posibilidad de
exacerbar una infección.
EVOLUCIÓN
Se inició una reducción gradual de prednisona. Los
hemocultivos para micobacterias permanecieron negativos después de 42 días de
incubación.
Cuatro meses más tarde, desarrolló dolor torácico
pleurítico, disnea de esfuerzo y ortopnea. Sus calambres abdominales se habían
resuelto, pero continuaban las fiebres intermitentes. Tomaba prednisona a dosis
de 10 mg diarios. La ecocardiografía transtorácica ambulatoria mostró función
biventricular y valvular normal. La angiografía por TC de tórax mostró derrame
pleural en el pulmón derecho sin embolia pulmonar. Un panel completo de genes
autoinflamatorios sugirió una variante de alto riesgo en el gen que codifica el
dominio de oligomerización de unión a nucleótidos que contiene 2 ( NOD2 ).
Debido a que este hallazgo es característico del síndrome de Yao, una
enfermedad autoinflamatoria, se inició un ensayo con sulfasalazina, en espera
de pruebas genéticas confirmatorias.
PONENTE
El síndrome de Yao es una enfermedad autoinflamatoria
asociada con la desregulación del sistema inmunitario innato. El diagnóstico de
la afección requiere criterios clínicos (al menos dos episodios periódicos de
fiebre, dermatitis o ambos, además de al menos uno de los siguientes síntomas o
signos: dolor abdominal, diarrea, síntomas de síndrome sicca, pericarditis o
pleuritis), criterios moleculares (presencia de la variante IVS8 +158 de NOD2
de alto riesgo [encontrada en aproximadamente el 15 % de las personas judías
asquenazíes sanas] o R702W [encontrada en aproximadamente el 4,3 % de las
personas blancas sanas]), además de un conjunto específico de criterios de
exclusión. Aunque este paciente cumplió con estos criterios, no tenía
dermatitis, que ocurre en la mayoría de los casos. Análisis genético confirmatorio
de NOD2 asociado al síndrome de variantes Yao, aún estaba pendiente. En este
contexto, hubiera sido razonable diferir el tratamiento con sulfasalazina a la
espera de más información.
EVOLUCIÓN
Los síntomas respiratorios del paciente continuaron
progresando. Se presentó a una clínica ambulatoria de nuestra institución para
otra opinión. Había dejado de tomar sulfasalazina. Había continuado con la
reducción gradual de prednisona y estaba tomando 7 mg de prednisona al día.
Refería fatiga, fiebre, anorexia, sudores nocturnos, tos seca, disnea de
esfuerzo, ortopnea, dolor torácico pleurítico, estreñimiento y artralgias sin
tumefacción articular ni dolor abdominal. Había perdido 22,7 kg desde el inicio
de la poliartritis migratoria. Al examen físico, la frecuencia respiratoria era
de 24 respiraciones por minuto y la temperatura de 36,7°C; crepitantes
inspiratorios, presión venosa yugular elevada y edema con fóvea +1 en ambas
piernas. No había linfadenopatía palpable, exantema, tumefacción articular,
eritema o déficits neurológicos. Los resultados del análisis genético fueron
positivos para el alelo NOD2 IVS8+158.
La TAC de tórax sin contraste mostró engrosamiento
pleural bilateral con derrame moderado en el pulmón derecho, derrame
pericárdico y adenopatías mediastínicas y subcarinales ( Figura 1 ).

Figura 1. TC de tórax mostrando engrosamiento pleural
y derrame pleural.
En el panel A (corte axial), y en el panel B (corte
coronal), las imágenes de tomografía de la parte inferior del tórax sin
contraste, muestran derrame pericárdico (flecha sólida), y engrosamiento
pleural (flecha discontinua), así como derrame pleural (asterisco), en el
pulmón derecho.
La ecocardiografía transtorácica mostró engrosamiento
pericárdico con trazas de derrame y características fisiológicas constrictivas
(desviación del tabique ventricular respiratorio, variación respiratoria en el
Doppler de entrada mitral >25% e inversiones diastólicas espiratorias en la
vena hepática). FDG-PET-CT mostró captación lineal leve de FDG dentro de las
superficies pleural y pericárdica, así como linfadenopatía torácica interna y
mediastínica ávida de FDG ( Figura 2).
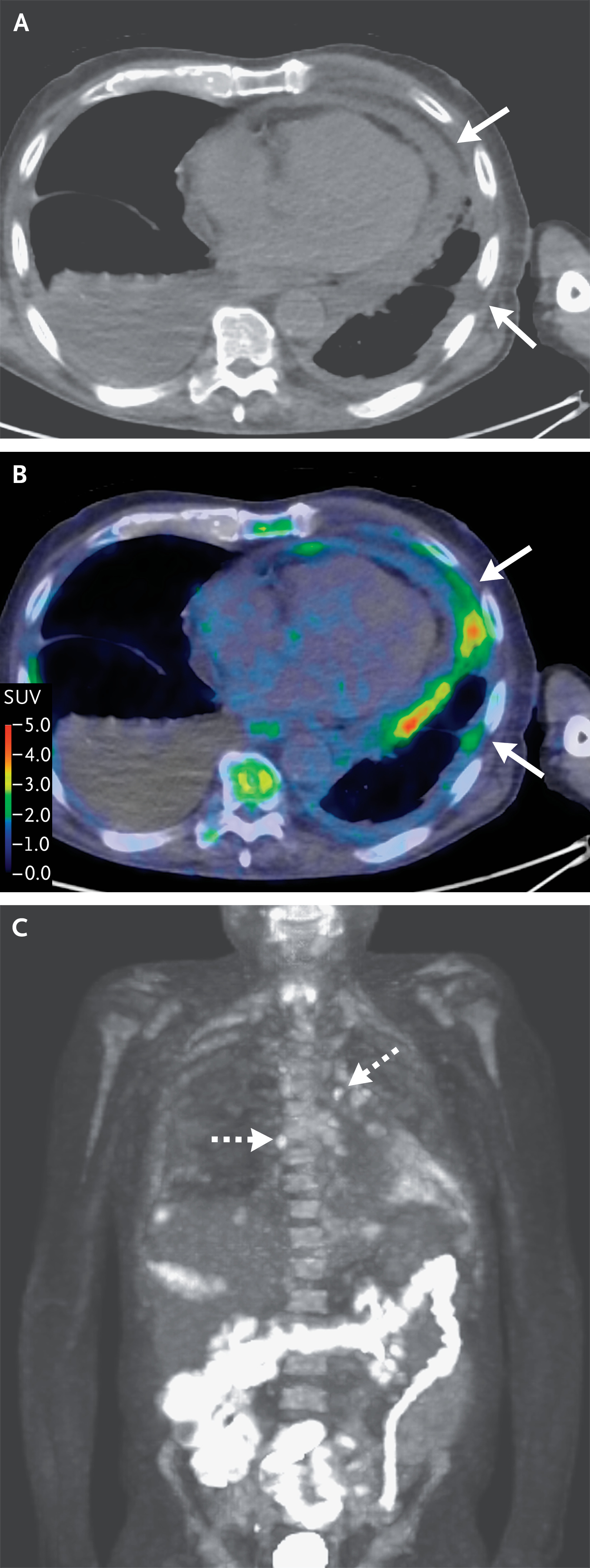
Figura 2. Imágenes FDG-PET-CT que muestran derrames
pleurales y pericárdicos ávidos de FDG.
Una imagen de TC de dosis baja sin material de
contraste (Panel A) y una tomografía por emisión de positrones (PET) fusionada
con 18F-fluorodesoxiglucosa (FDG) y una imagen de TC (Panel B) a través de la
parte inferior del tórax y una FDG-PET de proyección de máxima intensidad.
-imagen corporal (Panel C) muestra engrosamiento pleural y pericárdico con
avidez moderada de FDG (flechas sólidas; valor de captación estandarizado
máximo [SUV], 4,2) y ganglios linfáticos torácicos internos y mediastínicos
ávidos de FDG (flechas discontinuas; SUV máximo, 2,9).
Se ordenó un análisis de sangre que mide el ADN libre
de células microbianas (cfDNA) de más de 1000 especies diferentes de patógenos
muertos, moribundos o vivos (Prueba de Karius) debido a la sospecha continua de
una causa infecciosa. La resonancia magnética cardíaca mostró hallazgos
compatibles con miopericarditis con función y tamaño biventricular normales (
Figura 3 ). Durante la evaluación ambulatoria, se desarrolló un empeoramiento
de la disnea y la ortopnea, y el paciente fue ingresado en el hospital.

Figura 3. Resonancia magnética cardíaca que muestra
miopericarditis.
Una imagen con saturación de grasa ponderada en T2 del
miocardio basal en la proyección axial corta muestra una hiperintensidad T2
sutil y parcheada, más marcada en los segmentos inferior y anteroseptal (flecha
continua). Se evidencia derrame pericárdico (asterisco) y engrosamiento
pericárdico (flechas discontinuas). Las imágenes de eje corto realzadas con
contraste de gadolinio retardado a través del miocardio basal (Panel B)
muestran realce retardado parcheado en los segmentos inferior e inferolateral
(puntas de flecha) con realce del pericardio (flechas discontinuas). Estos
hallazgos son consistentes con miopericarditis.
PONENTE
Ahora se ha desarrollado pleuritis y miopericarditis.
La linfadenopatía ávida de FDG podría ser reactiva, aunque sigue siendo posible
el cáncer (con metástasis pleural y pericárdica). El siguiente paso sería
realizar una toracocentesis diagnóstica y terapéutica. Los resultados de una prueba
de cfDNA pueden ser útiles antes de que se intensifique la inmunosupresión,
pero esta prueba no sustituye a las pruebas validadas de infección, como la
PCR.
EVOLUCIÓN
Se inició tratamiento con furosemida intravenosa y se
aumentó la dosis de prednisona a 40 mg diarios. Una ecografía pleural mostró un
derrame pleural multiloculado en el pulmón derecho, por lo que se colocó un
tubo torácico. El nivel de lactato deshidrogenasa en líquido pleural fue de 111
U por litro (nivel de lactato deshidrogenasa en suero, 226 U por litro [valor
de referencia, <222]), el nivel de proteína total en líquido pleural fue de
3,6 g por decilitro (nivel de proteína total en suero, 6,3 g por decilitro), el
pH 7,37 y el recuento de glóbulos blancos 4463 por microlitro (100% linfocitos).
Dada la disnea progresiva y la presencia del NOD2 IVS8 +158alelo, se administró
canakinumab (un antagonista del receptor de la interleucina-1) para el presunto
síndrome de Yao después de que los cultivos bacterianos del líquido pleural
hubieran sido negativos durante 48 horas. Luego, una prueba de cfDNA dio
positivo para 73 moléculas de ADN de T. whipplei por microlitro (valor de
referencia, <10).
PONENTE
Los resultados de las pruebas de líquido pleural son
consistentes con un derrame exudativo (proporción de proteína en líquido
pleural a proteína en suero de >0.5). En el contexto de múltiples
manifestaciones consistentes con la enfermedad de Whipple (incluyendo serositis
con derrame exudativo, poliartritis migratoria prodrómica, calambres abdominales,
pérdida de peso y deficiencia de hierro), la prueba positiva de cfDNA sugiere
fuertemente este diagnóstico, aunque esta prueba no es diagnóstica ni se
recomienda actualmente. en diagnóstico. Los criterios diagnósticos sugeridos
para la enfermedad de Whipple incluyen presentación patognomónica de miorritmia
oculomasticatoria u oculofacial-esquelética (es decir, convergencia rítmica de
los ojos y contracciones sincrónicas de los músculos masticatorios y los
músculos esqueléticos proximales y distales), Macrófagos PAS positivos con
tinción Ziehl-Nielsen negativa en una muestra de biopsia de intestino delgado
en pacientes con síntomas gastrointestinales predominantes y al menos dos
pruebas positivas diferentes (es decir, tinción PAS, PCR o tinción inmunohistoquímica)
de la misma muestra o una prueba positiva de al menos dos muestras diferentes.
Por lo tanto, el diagnóstico debe confirmarse realizando PCR para T. whipplei,
tinción PAS o pruebas inmunohistoquímicas de líquido pleural y muestras de
biopsia de intestino delgado. Si se diagnostica la enfermedad de Whipple,
también se debe realizar una prueba de PCR de T. whipplei en el líquido
cefalorraquídeo (LCR) independientemente de los síntomas o signos neurológicos
para evaluar la participación del sistema nervioso central (SNC).
EVOLUCIÓN
El paciente no tenía déficits neurológicos focales,
amnesia, confusión o ataxia. Un examen fue negativo para miorritmia
oculomasticatoria u oculo-facial-esquelética. Se inició tratamiento con
ceftriaxona intravenosa a dosis de 2 g diarios, ante la alta sospecha de
enfermedad de Whipple. Se redujo la dosis de prednisona. Una repetición de la
prueba del VIH fue negativa. La PCR para T. whipplei del líquido pleural fue
positiva. Las biopsias duodenales endoscópicas repetidas mostraron macrófagos
positivos para PAS en el duodeno, y la prueba de PCR para T. whipplei fue
positiva ( Figura 4 ), lo que confirma el diagnóstico de la enfermedad de
Whipple. Los resultados de la prueba PCR de T. whipplei en el LCR fueron
negativos.

Figura 4. Muestra de biopsia duodenal que muestra
macrófagos positivos para PAS.
Una muestra de biopsia duodenal muestra vellosidades
aplanadas (asterisco) y la tinción con ácido peryódico de Schiff (PAS) revela
numerosos macrófagos teñidos de rojo intenso con PAS positivo (flecha), lo que
indica la presencia de Tropheryma whipplei .
En el seguimiento ambulatorio 6 semanas después,
informó una marcada disminución de sus síntomas respiratorios y
constitucionales. Ya había completado un ciclo de ceftriaxona de 2 semanas y se
inició un ciclo de trimetoprima-sulfametoxazol de 12 meses. Estaba tomando 5 mg
de prednisona al día y la dosis se estaba reduciendo gradualmente.
PONENTE
La enfermedad de Whipple se diagnosticó y trató
adecuadamente, con una marcada mejoría en el seguimiento a corto plazo. Se
necesitará un seguimiento estrecho para asegurar la resolución de los síntomas
y la erradicación de T. whipplei . Si los síntomas persistieran a pesar de la
erradicación documentada, se debe reconsiderar el síndrome de Yao y tratarlo
con la inmunosupresión adecuada.
COMENTARIO
Este paciente presentaba poliartritis migratoria
seronegativa refractaria al tratamiento con prednisona y metotrexato. La
poliartritis fue seguida por síntomas constitucionales, calambres abdominales,
estreñimiento y pérdida de peso. Se consideró la enfermedad de Whipple, pero el
diagnóstico se descartó prematuramente después de una tinción PAS negativa en
un número incierto de muestras de biopsia duodenal. El desarrollo subsiguiente
de serositis y síntomas respiratorios rápidamente progresivos y la detección de
T. whipplei en la prueba de Karius llevaron al diagnóstico de la enfermedad de
Whipple mediante pruebas de PCR en líquido pleural y muestras de biopsia de
intestino delgado.
La enfermedad de Whipple es un trastorno
multisistémico raro causado por T. whipplei , una actinobacteria intracelular
grampositiva en forma de bastón que se encuentra en el entorno natural y de la
cual los seres humanos son el único huésped conocido. 1 La tasa de incidencia
es de aproximadamente 1 a 6 casos por 10.000.000 de personas por año en todo el
mundo. 2 Un estudio poblacional reciente en los Estados Unidos reportó una
mayor prevalencia entre las personas blancas que entre las personas negras,
entre las personas no hispanas que entre las personas hispanas, y entre las
personas mayores de 65 años que entre las que tenían 65 años o más jóvenes,
pero no se encontraron diferencias materiales según el sexo. 2
La transmisión de T. whipplei ocurre a través del
contacto con suelo o aguas residuales contaminadas o por vía fecal-oral. 1,3
Una vez que una persona está infectada, T. whipplei reside dentro de las
células mononucleares de la sangre periférica o en las vacuolas de los
macrófagos en el intestino delgado, donde evade la inmunidad del huésped
creando un entorno antiinflamatorio. 1,4 La mayoría de las personas infectadas
finalmente eliminan la infección, algunas (<25 %) se convierten en
portadores asintomáticos a largo plazo y una pequeña minoría (<0,01 %) presenta
la enfermedad de Whipple clásica o localizada. 3,5El hallazgo de haplo insuficiencia
del factor regulador de interferón 4 en miembros de una familia que tenía la
enfermedad de Whipple y no en miembros de la familia no infectados sugiere que
la susceptibilidad genética puede ser la base del desarrollo de la enfermedad
sintomática en algunas personas, en particular las de ascendencia europea. 5
La enfermedad de Whipple clásica afecta
predominantemente al intestino delgado y las manifestaciones de presentación
comunes en series de casos incluyen pérdida de peso (90 %), diarrea por
malabsorción (75 %), dolor abdominal (60 %), artropatía migratoria seronegativa
no destructiva (85 %), fiebre (45%) y linfadenopatía (45%); el derrame pleural
ocurre con menos frecuencia y el linfoma extraintestinal ocurre en casos raros.
1,6,7 En la mayoría de los pacientes con enfermedad de Whipple clásica, la
artralgia ocurre como síntoma prodrómico años antes del inicio de otras
manifestaciones. 1 La afección a menudo se diagnostica incorrectamente como
artritis inflamatoria seronegativa, pero los síntomas no responden a la
inmunosupresión e incluso pueden empeorar con ella. 8Este fue el caso de
nuestro paciente, en quien desarrolló síntomas sistémicos, gastrointestinales y
respiratorios mientras tomaba prednisona. 9 En la enfermedad de Whipple
localizada, los órganos extraintestinales como el cerebro, el corazón, los
pulmones o las articulaciones se ven afectados sin afectación gastrointestinal
evidente. 3
El diagnóstico de la enfermedad de Whipple requiere un
alto índice de sospecha, lo que puede ser un desafío dada su rareza. La
infección por T. whipplei se puede diagnosticar mediante PCR o evaluación
histológica y tinción PAS de biopsias duodenales, fluidos corporales o tejidos
afectados. 3,4 En un pequeño estudio que involucró a pacientes con síntomas
predominantemente gastrointestinales, se informó que las heces y la saliva con
PCR positivo tenían una sensibilidad muy alta y un valor predictivo positivo,
10 pero se necesitan más datos para informar la utilidad de estas pruebas en el
diagnóstico de la enfermedad de Whipple. enfermedad. Por lo tanto, se deben
realizar PCR de confirmación, tinción de PAS y pruebas inmunohistoquímicas de
múltiples muestras de biopsia duodenal para confirmar el diagnóstico. 3,4Se ha
informado que la prueba de PCR de muestras de biopsia de intestino delgado en
la enfermedad de Whipple clásica tiene una sensibilidad de aproximadamente el
89 %, en comparación con aproximadamente el 60 % para la enfermedad de Whipple
localizada. 8 La prueba de T. whipplei por medio de PCR en líquido o tejido de
sitios extraintestinales afectados puede estar justificada si la prueba de
muestras de biopsia duodenal no es confirmatoria o es negativa en el contexto
de alta sospecha clínica de enfermedad de Whipple. 3,4
La secuenciación de próxima generación de cfDNA
microbiano se puede utilizar para evaluar las causas infecciosas de endocarditis
con hemocultivo negativo y antes de la intensificación de la inmunosupresión en
pacientes con presentaciones atípicas de enfermedad reumatológica. En el
presente caso, los resultados sugirieron el diagnóstico de enfermedad de
Whipple. Sin embargo, faltan datos para comparar las pruebas de cfDNA con las
pruebas de PCR convencionales, y actualmente no se recomienda el uso de la
prueba en pacientes con sospecha de enfermedad de Whipple.
El régimen de tratamiento más utilizado consiste en la
inducción con ceftriaxona o meropenem durante 2 semanas, seguida de
trimetoprima-sulfametoxazol durante 12 meses. 6 Un ensayo aleatorizado que
involucró a 40 pacientes asignados a recibir ceftriaxona o meropenem informó
una remisión clínica y de laboratorio en todos los pacientes de ambos grupos a
los 3 meses, que se mantuvo durante una mediana de seguimiento de 89 meses. 6
La remisión de laboratorio no requirió pruebas de PCR repetidas negativas. Sin
embargo, las pruebas repetidas después del tratamiento mostraron resultados
negativos de PCR duodenal en 18 de 19 pacientes y resultados negativos de PCR
en LCR inicialmente en los 10 pacientes con enfermedad del SNC. (Más tarde, se
observó que un paciente tenía LCR positivo en la prueba de PCR repetida en
ausencia de síntomas, que finalmente desapareció con un tratamiento adicional).
Traduccción de:
Jwan A. Naser, M.B., B.S., Delvise Fogwe, M.D., Matthew Ho, M.B., B.Ch., B.A.O., Ph.D., Cristina Corsini Campioli, M.D., and Aditya Shah, M.B., B.S.
NEJM
References
1. Marth T, Moos V, Müller C, Biagi F,
Schneider T. Tropheryma whipplei infection and
Whipple’s disease. Lancet Infect
Dis 2016;16(3):e13-e22.
2. Elchert JA, Mansoor E, Abou-Saleh M,
Cooper GS. Epidemiology of Whipple’s
disease in the USA between 2012 and
2017: a population-based national study.
Dig Dis Sci 2019;64:1305-11.
3. Dolmans RA, Boel CH, Lacle MM,
Kusters JG. Clinical manifestations, treatment, and
diagnosis of tropheryma whipplei infections. Clin Microbiol Rev 2017;
30:529-55.
4. Schneider T, Moos V, Loddenkemper C,
Marth T, Fenollar F, Raoult D. Whipple’s
disease: new aspects of pathogenesis and
treatment. Lancet Infect Dis 2008;8:179-90.
5. Guérin A, Kerner G, Marr N, et al.
IRF4 haploinsufficiency in a family with
Whipple’s disease. Elife 2018;7:e32340.
6. Feurle GE, Junga NS, Marth T. Efficacy
of ceftriaxone or meropenem as initial
therapies in Whipple’s disease. Gastroenterology
2010;138:478-86.
7. Gillen CD, Coddington R, Monteith
PG, Taylor RH. Extraintestinal lymphoma
in association with Whipple’s disease. Gut
1993;34:1627-9.
8. Tison A, Preuss P, Leleu C, et al.
Rheumatological features of Whipple disease. Sci Rep
2021;11:12278.
9. Marth T. Systematic review: Whipple’s
disease (Tropheryma whipplei infection)
and its unmasking by tumour necrosis
factor inhibitors. Aliment Pharmacol Ther
2015;41:709-24.
10. Fenollar F, Laouira S, Lepidi H, Rolain
J-M, Raoult D. Value of Tropheryma whipplei
quantitative polymerase chain reaction assay for the diagnosis of Whipple
disease: usefulness of saliva and stool
specimens for first-line screening. Clin
Infect Dis 2008;47:659-67.



