Mujer de 86 años con diagnostico de hipertension arterial en tratamiento con hidroclorotiazida 12.5 mg/dia y Aspirina de 80 mg/dia hace 12 meses. Hace 3 meses presenta en ambas piernas unas lesiones dermicas maculares marrón-rojizas que tienden a coalescer y han formado una zona de piel acartonada y rojiza en la parte posteior de la pierna. Se suspendió la aspirina hace 3 meses sin mejoria en las lesiones que continuaron apareciendo. Tiene hemograma y perfil hepatico normal con plaquetas de207 mil/ul y perfil de coagulación normal. Ha sido evaluada por cardiologia, geriatria y hematología sin llegar a un diagnóstico. En los resultados de laboratorio se encontró factor reumatoideo positivo y la biopsia de piel mostró Vasculopatía Livedoide (atrofia blanca).

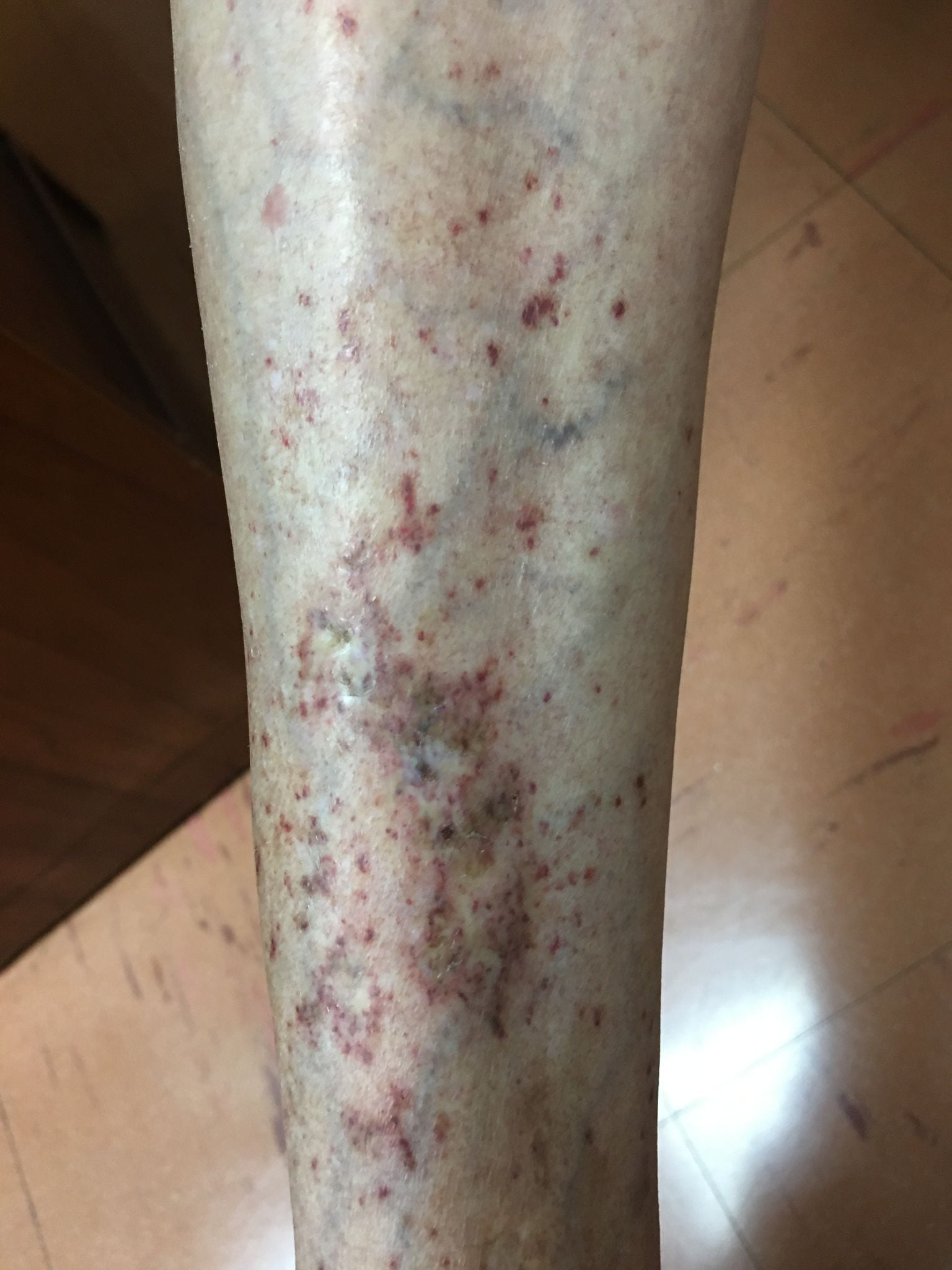



Lamentablemente la paciente no volvió a consulta
perdiéndose en el seguimiento.

Gentileza:
Dr. Martín Vásquez.
Médico. Trujillo Perú.
VASCULOPATÍA LIVEDOIDE
INTRODUCCIÓN
La vasculopatía livedoide
es una vasculopatía cutánea trombo-oclusiva, dolorosa y crónica que afecta a la
parte distal de las extremidades inferiores y los pies. Las características
clínicas características incluyen cambios en la piel livedoide (nódulos
eritematosos lineales o angulares), atrofia blanca (placas lisas de color
blanco marfil) y ulceración. El diagnóstico se confirma mediante una biopsia de
piel que demuestra anormalidades vasculares características, que incluyen
trombosis intraluminal, proliferación endotelial y degeneración hialina
subintimal.
La patogenia de la
vasculopatía livedoide no está clara. El trastorno puede ocurrir de forma
independiente o en asociación con trombofilia adquirida o hereditaria o varias
enfermedades sistémicas.
NOMENCLATURA
La literatura sobre la
vasculopatía livedoide es muy problemática con respecto a la nomenclatura [ 1
]. Se ha utilizado una variedad de otros términos para referirse a esta afección,
con mayor frecuencia "atrofia blanca" y "vasculitis livedoide (o
livedo)". Sin embargo, "atrofia blanca" se usa más
apropiadamente como un término descriptivo para las placas lisas de color
blanco marfil que se presentan de manera característica en la vasculopatía
livedoide, pero que también pueden ocurrir en otros trastornos, en particular
la insuficiencia venosa. Debe evitarse el uso de "vasculitis" porque
la vasculitis verdadera, como se demuestra por la inflamación destructiva
dentro de la pared del vaso sanguíneo, está ausente.
Los términos menos
comunes que se han utilizado para referirse a la vasculopatía livedoide en la
literatura incluyen vasculopatía hialinizante segmentaria, livedo reticularis
con ulceraciones de verano, livedo reticularis con ulceraciones de invierno y
úlceras purpúricas dolorosas con patrón reticular en las extremidades
inferiores, o painful purpuric ulcers with reticular patterning on the lower
extremities (PURPLE).
EPIDEMIOLOGÍA Y PATOGENIA
La vasculopatía livedoide
ocurre principalmente en adultos jóvenes a de mediana edad y es más común en
mujeres que en hombres. La patogenia no se conoce bien, pero se postula que
implica un aumento de la coagulación o una alteración de la fibrinólisis que
provoca la oclusión de los vasos sanguíneos dérmicos con trombos de fibrina [ 2
].
CARACTERÍSTICAS CLÍNICAS
La vasculopatía livedoide
ocurre en la parte inferior de la pierna (el sitio más común), el tobillo y/o
la parte dorsal del pie [ 3 ]. La presentación suele ser bilateral, pero también
puede ocurrir afectación unilateral. Los hallazgos clínicos característicos son
cambios livedoides y atrofia blanca. La ulceración es un hallazgo común
adicional.
- Cambios livedoides: los cambios livedoides son máculas, pápulas o nódulos profundos, apenas palpables y ligeramente eritematosos que tienen una forma lineal o angular. La apariencia se asemeja a la livedo reticularis parcheada ( imagen 6 ).

Imagen 6. Vasculopatía
livedoide.
Parches, patrón incompleto
similar a livedo reticularis en los pies y atrofia blanca, telangiectasias y
úlceras estrelladas en la parte inferior de la pierna.
- Atrofia blanca: la atrofia blanca aparece como placas atróficas lisas de color blanco marfil rodeadas de hiperpigmentación y telangiectasias ( imagen 7,8,9 ). Las placas a menudo tienen pequeñas cantidades de pigmento punteado. La atrofia blanca puede desarrollarse en sitios de ulceración previa o en ausencia de úlceras previas.

Imagen 7. Atrofia blanca.
Cicatrices atróficas,
blancas, estrelladas consistentes con atrofia blanca están presentes en la
extremidad inferior en este paciente con vasculitis livedoide.

Imagen 8. Atrofia blanca.
Cicatrices atróficas,
blancas, estrelladas consistentes con atrofia blanca están presentes en la
extremidad inferior en este paciente con vasculitis livedoide.

Imagen 9. Vasculopatía livedoide.
Áreas de color blanco
marfil, similares a placas, hiperpigmentación y ulceración.
- Úlceras : las úlceras superficiales son comunes y generalmente varían de 1 a 5 mm de diámetro ( imagen 10,11). También pueden ocurrir úlceras más grandes y profundas. La eliminación de la escara revelará una úlcera en sacabocados, angular o estrellada.

Imagen 10. Vasculopatía
livedoide.
Úlceras estrelladas
"en sacabocados" en un paciente con vasculopatía livedoide.

Úlceras superficiales
sobre el dorso de los pies con hiperpigmentación posinflamatoria y
cicatrización estrellada.
Los cambios livedoides
son a menudo la manifestación clínica inicial de la vasculopatía livedoide.
Alternativamente, el edema del tobillo o pequeñas pápulas ligeramente elevadas
con telangiectasias o púrpura y petequias periféricas pueden preceder a los
cambios livedoides. Las úlceras suelen ser persistentes; incluso con
tratamiento, la curación puede tardar varios meses.
La mayoría de los
pacientes con vasculopatía livedoide experimentan dolor considerable, ardor o
picazón en las áreas afectadas.
PATOLOGÍA
Los cambios patológicos
característicos ocurren en los vasos sanguíneos de la dermis superior, media
y/o inferior. Los vasos sanguíneos muestran engrosamiento y trombosis focal,
con proliferación endotelial y degeneración hialina de la capa subintimal [ 4 ].
El material hialino se tiñe positivamente con la técnica de ácido peryódico de
Schiff, pero también se visualiza bien con la tinción de hematoxilina eosina (
imagen 12 ).

Imagen 12. Vasculopatía
livedoide.
Vasculopatía hialinizante
segmentaria. Cambios hialinizados dentro de la capa subintimal de las paredes
de los vasos sanguíneos y trombosis vascular en la dermis superficial y
profunda.
Las láminas elásticas de
los vasos afectados suelen conservarse y la pared vascular rara vez se
destruye. La luz de los vasos están ocluidas por la proliferación de células
incrustadas en material fibrinoide suelto. La reacción inflamatoria circundante
es relativamente leve y consiste principalmente en linfocitos. Puede haber
extravasación de glóbulos rojos.
TRASTORNOS ASOCIADOS
La vasculopatía livedoide
puede ocurrir con o sin factores de riesgo predisponentes identificables para
la trombosis. Los ejemplos de factores de riesgo de trombosis que se han
relacionado con la vasculopatía livedoide incluyen anticuerpos
antifosfolípidos, paraproteinemias y trastornos genéticos que predisponen a
trombosis.
La vasculopatía livedoide
también puede ocurrir en el contexto de una enfermedad definida,
particularmente una enfermedad reumática sistémica. Las enfermedades más
comúnmente diagnosticadas en el contexto de la vasculopatía livedoide son el
síndrome antifosfolípido primario, el lupus eritematoso sistémico, la artritis
reumatoide, la esclerosis sistémica, la enfermedad mixta del tejido conectivo y
la enfermedad indiferenciada del tejido conectivo [ 3,5,6 ].
DIAGNÓSTICO
El diagnóstico de
vasculopatía livedoide se confirma mediante la detección de hallazgos
histológicos consistentes en un paciente con hallazgos físicos sugestivos (p.
ej., cambios livedoides, atrofia blanca, úlceras) en las extremidades
inferiores. Las manifestaciones cutáneas por sí solas son insuficientes para
confirmar el diagnóstico, ya que pueden ocurrir hallazgos similares en otros
trastornos. Por lo tanto, una biopsia de piel es obligatoria.
Para los pacientes con
úlceras, una historia clínica y un examen físico cuidadosos pueden ser útiles
para determinar si se deben incluir otros diagnósticos en el diagnóstico
diferencial. El enfoque del diagnóstico diferencial de las úlceras en las
piernas se revisa por separado.
El tipo de biopsia ideal
es una biopsia incisional fusiforme que incluye grasa subcutánea. Como
alternativa, se puede realizar una biopsia con sacabocados de 4 a 6 mm [ 2 ].
El sitio óptimo para una biopsia es el borde de una nueva úlcera o una nueva
pápula purpúrica. Los hallazgos histológicos en la vasculatura dérmica que
respaldan firmemente el diagnóstico de vasculopatía livedoide son [ 2 ]:
- Proliferación endotelial
- Degeneración hialina subintimal
DIAGNÓSTICO DIFERENCIAL
El diagnóstico
diferencial de la vasculopatía livedoide incluye una amplia gama de condiciones
que pueden causar decoloración de la piel, úlceras cutáneas o nódulos en las
extremidades inferiores. Los trastornos más comunes en el diagnóstico
diferencial son la enfermedad venosa crónica de las extremidades inferiores, la
enfermedad vascular periférica y la vasculitis.
●Enfermedad venosa
crónica: la insuficiencia venosa que afecta a las extremidades inferiores a
menudo se presenta con hiperpigmentación cutánea, edema y varicosidades en las
extremidades inferiores. La ulceración puede ocurrir y se encuentra con mayor
frecuencia cerca del maléolo medial. Las úlceras suelen ser superficiales con
bordes irregulares y exudado fibrinoso amarillo ( imagen 13 ). También puede
ocurrir atrofia blanca. El diagnóstico a menudo se puede hacer clínicamente; la
ecografía venosa puede ser útil cuando el diagnóstico es incierto.

Imagen 13. Dermatitis por
estasis.
Dermatitis por estasis
que rodea las úlceras por insuficiencia venosa.
●Enfermedad vascular
periférica : la enfermedad vascular periférica de las extremidades inferiores
puede provocar úlceras isquémicas, que a menudo se encuentran en los dedos de
los pies, el talón, el maléolo o la espinilla. Las úlceras dolorosas están bien
delimitadas, con aspecto de "sacado" ( foto 14 ). Los pulsos
periféricos están disminuidos. El diagnóstico se puede corroborar mediante la
prueba del índice tobillo-brazo.

Imagen 14. Úlcera
isquémica.
Úlceras "en
sacabocados" bien delimitadas con escara en la extremidad inferior.
Vasculitis: la vasculitis
que involucra vasos sanguíneos cutáneos de tamaño mediano (p. ej.,
poliarteritis nodosa cutánea, vasculitis asociada a anticuerpos anticitoplasma
de neutrófilos [ANCA], crioglobulinemia mixta, tromboangeítis obliterante,
arteritis trombofílica linfocítica) puede presentarse con nódulos subcutáneos,
úlceras, livedo reticularis o livedo racemosa. Una biopsia de piel puede
diferenciar la vasculopatía livedoide de la vasculitis.
Los ejemplos de otros
trastornos en el diagnóstico diferencial incluyen pioderma gangrenoso, lesiones
cutáneas traumáticas o facticias y las diversas causas adicionales de úlceras
en las piernas.
EVALUACIÓN
ADICIONAL
Después de que se haya
confirmado histológicamente el diagnóstico de vasculopatía livedoide, se debe
evaluar a los pacientes para detectar trastornos subyacentes. La evaluación
debe comenzar con una historia clínica completa, una revisión de los sistemas y
un examen físico para evaluar los hallazgos que sugieran trombofilia o una
enfermedad reumática sistémica. Aunque generalmente realizamos pruebas de
laboratorio para trombofilia en todos los pacientes, limitamos la investigación
de laboratorio para enfermedades reumáticas sistémicas a pacientes que muestran
otros hallazgos que sugieren tales enfermedades.
Los ejemplos de estudios
que a menudo se incluyen en nuestra evaluación de laboratorio incluyen:
- Pruebas para trombofilia adquirida y hereditaria
- Hemograma completo
- Panel metabólico completo
- Análisis de orina
- Velocidad de sedimentación globular y proteína C reactiva
- Factor reumatoideo y anticuerpos contra péptidos citrulinados cíclicos
- Niveles de complemento sérico (p. ej., CH50, C3 y C4)
- Título de anticuerpos antinucleares
- Anticuerpos anticitoplasma de neutrófilos (ANCA)
- Crioglobulinas séricas
- Electroforesis e inmunofijación de proteínas séricas
El valor principal en la
evaluación de pacientes para una enfermedad reumática sistémica es la
identificación de pacientes en riesgo de otras complicaciones de la condición
identificada. El tratamiento de la enfermedad reumática sistémica subyacente no
parece tener un efecto constante sobre la vasculopatía livedoide, y la
presencia de una enfermedad reumática sistémica no suele alterar el enfoque del
tratamiento. Por el contrario, la presencia de trombofilia influye en nuestro enfoque
del tratamiento.
TRATAMIENTO
El tratamiento de la
vasculopatía livedoide suele implicar una combinación de intervenciones. Ningún
enfoque terapéutico único es efectivo para todos los pacientes [ 7 ].
Medidas generales: el manejo del dolor y el cuidado de las
heridas son componentes importantes del manejo. Dejar de fumar y la compresión
también pueden ser beneficiosos.
Manejo del dolor: el
dolor secundario a la vasculopatía livedoide puede ser intenso. Nuestro enfoque
de primera línea para el manejo del dolor generalmente consiste en acetaminofén
o medicamentos antiinflamatorios no esteroideos, como la indometacina. Los
tratamientos típicamente utilizados para el dolor neuropático, como los
fármacos tricíclicos, la gabapentina , la pregabalina o la carbamazepina ,
pueden ser útiles para los pacientes con ulceraciones persistentes [ 2 ].
Cuidado de heridas: el
cuidado de heridas debe implicar el mantenimiento de un ambiente húmedo para la
herida y el control de la sobreinfección. Los principios generales del
tratamiento de heridas se revisan por separado.
Dejar de fumar: aunque
no se han estudiado los efectos de dejar de fumar en la vasculopatía livedoide,
alentamos a los pacientes a que dejen de fumar debido a los efectos negativos
del tabaquismo en la cicatrización de heridas. Además, existe una relación
propuesta entre la vasculopatía livedoide y el tabaquismo. En una serie de 45
pacientes con vasculopatía livedoide evaluados en un centro de atención
terciaria en los Estados Unidos, el 47 por ciento informó antecedentes de tabaquismo
[ 3 ].
Compresión: la
experiencia clínica sugiere que la terapia de compresión es beneficiosa en
pacientes con insuficiencia venosa asociada, siempre que los resultados del
índice tobillo-brazo no sugieran insuficiencia arterial concomitante [ 2 ]. La
mejora puede estar relacionada con un efecto estimulante de la compresión sobre
la actividad fibrinolítica [ 8 ].
Terapia farmacológica: los datos limitados sugieren que las terapias
que minimizan el riesgo de trombosis, incluidos los agentes antiplaquetarios,
anticoagulantes y fibrinolíticos, pueden mejorar la vasculopatía livedoide [
2,9 ]. También se ha informado el beneficio de las intervenciones
inmunomoduladoras, vasodilatadoras y de otro tipo. La terapia de combinación es
a menudo necesaria.
Nuestro enfoque: ningún ensayo aleatorio ha evaluado las
terapias para la vasculopatía livedoide, y el mejor enfoque para el tratamiento
no está claro. Nuestro enfoque se revisa aquí. Otros enfoques pueden ser
razonables.
El agente antiplaquetario
aspirina, un agente oral generalmente bien tolerado y ampliamente disponible,
es nuestra terapia de primera línea preferida para pacientes sin trombofilia
identificada. Por lo general, recetamos aspirina sola para la terapia inicial,
aunque algunos médicos usan la terapia combinada con dipiridamol. La
pentoxifilina es nuestro tratamiento inicial favorito para los pacientes que no
toleran la aspirina. Para los pacientes que pueden tolerar la aspirina pero no
mejoran con la aspirina sola, generalmente agregamos pentoxifilina.
Cuando los pacientes
tienen respuestas insuficientes a la aspirina y la pentoxifilina , a menudo se
procede a la anticoagulación. Por lo general, usamos warfarina, heparina de
bajo peso molecular (BPM) o un anticoagulante oral directo, como rivaroxabán .
La mayoría de los pacientes responden a uno de estos agentes. Para nuestros
raros pacientes con enfermedad refractaria, hemos tenido cierto éxito con la
terapia trombolítica tisular (activador tisular del plasminógeno).
Nuestro enfoque para los
pacientes con trombofilia identificada es diferente. Los anticoagulantes se
utilizan a menudo como tratamiento de primera línea; sin embargo, la selección
de un agente inicial debe basarse en el conocimiento de las terapias conocidas
para reducir la trombosis en la trombofilia específica.
No hay un criterio de
valoración fijo para la terapia en pacientes que responden al tratamiento. Se
puede intentar la interrupción del tratamiento después de la cicatrización de
la úlcera; sin embargo, puede ser necesario un tratamiento continuo para
mantener la mejoría.
Agentes anti plaquetarios: se ha informado una mejoría del tratamiento
con agentes antiplaquetarios, como aspirina , dipiridamol y pentoxifilina .
Aspirina con o sin
dipiridamol: los informes de casos y las
series de casos documentan los beneficios de la aspirina sola o en combinación
con el agente antiplaquetario dipiridamol [ 10-13 ]. La aspirina generalmente
se administra a pacientes adultos en una dosis de 325 a 365 mg por día; la
dosis típica de dipiridamol es de 75 mg cuatro veces al día. Se espera una
mejoría en los signos y síntomas de la vasculopatía livedoide dentro de los
primeros meses de tratamiento. Los efectos adversos de la aspirina se discuten
por separado.
Pentoxifilina: la pentoxifilina se puede administrar sola o
en combinación con aspirina . En particular, la pentoxifilina es un tratamiento
inicial favorable para los pacientes que no toleran la aspirina. La
pentoxifilina mejora la hiperviscosidad de la sangre y tiene efectos
inhibitorios sobre la agregación de plaquetas y eritrocitos. El fármaco ha
resultado útil para la vasculopatía livedoide en series de casos [ 14,15 ].
Los adultos pueden
tratarse con 400 mg de pentoxifilina de una a tres veces al día. Se espera una
mejoría clínica dentro de los primeros meses de tratamiento. El malestar
gastrointestinal es un posible efecto secundario.
Anticoagulantes: se ha informado una mejoría del tratamiento
con anticoagulantes, como warfarina , heparina y anticoagulantes orales
directos.
Warfarina o heparina: la rápida mejoría clínica durante el
tratamiento con warfarina oral o heparina de BPM subcutánea está documentada en
informes de casos y series de casos [ 9,16,17 ]. La warfarina se dosifica para
lograr un índice internacional normalizado (INR) entre 2 y 3 y puede conducir a
una mejoría notable dentro de los dos primeros meses de tratamiento [ 18-20 ].
La enoxaparina de BPM se puede administrar por vía subcutánea a razón de 1
mg/kg cada 12 horas durante seis meses, seguida de un tratamiento una vez al
día. Se ha producido una marcada mejoría clínica en los primeros meses de tratamiento
con heparina de bajo peso molecular [ 21,22]. Los requisitos de seguimiento y
los efectos adversos de la warfarina y la heparina de bajo peso molecular se
revisan en detalle por separado. La warfarina está contraindicada en el
embarazo.
Anticoagulantes orales directos: los anticoagulantes orales directos son
opciones más nuevas para la anticoagulación que no requieren el control
frecuente necesario para la terapia con warfarina y se pueden administrar por
vía oral, a diferencia de la terapia con heparina de bajo peso molecular. En un
estudio no controlado de 12 semanas, el tratamiento de 25 pacientes con
vasculopatía livedoide dolorosa con rivaroxabán oral (10 mg dos veces al día
con la opción de reducir a una dosis diaria al mejorar) se asoció con una
reducción del dolor, el eritema y la ulceración [ 23 ] . El beneficio de
rivaroxabán y otros anticoagulantes orales directos, incluidos apixabán ,
edoxabán y dabigatrán , está documentado en informes de casos o series de casos
[ 9,24-28]. Los riesgos de los anticoagulantes orales directos se analizan por
separado.
Otras terapias: otras terapias que pueden ser útiles según
informes de casos o series de casos pequeños incluyen el activador del
plasminógeno tisular (10 mg por vía intravenosa durante 14 días) [ 29-31 ],
doxiciclina (100 mg dos veces al día), danazol (200 mg por día ) [ 23,32 ],
oxígeno hiperbárico (1,5 a 2 horas para hasta 15 sesiones de tratamiento) [ 33
], inmunoglobulina intravenosa (infusiones mensuales de 0,5 g/kg administradas durante
dos o tres días consecutivos) [ 34 ], nifedipina ( 10 a 20 mg tres veces al
día) [ 35 ], becaplermina tópica [ 36], psoraleno más ultravioleta A (PUVA; 0,5
a 1 mJ/cm 2 por sesión de tratamiento) [ 37,38 ], sulfasalazina (1 g tres veces
al día) [ 39,40 ] y ablación endovenosa para la insuficiencia venosa asociada [
41 ].
PRONÓSTICO
La vasculopatía livedoide
recurre a menudo tras la interrupción del tratamiento. Suele ser necesario un
tratamiento a largo plazo con un régimen eficaz.
REFERENCES
Jorizzo JL. Livedoid vasculopathy: what is it? Arch
Dermatol 1998; 134:491.
Alavi A, Hafner J, Dutz JP, et al. Livedoid
vasculopathy: an in-depth analysis using a modified Delphi approach. J Am Acad
Dermatol 2013; 69:1033.
Hairston BR, Davis MD, Pittelkow MR, Ahmed I. Livedoid
vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol
2006; 142:1413.
Bard JW, Winkelmann RK. Livedo vasculitis. Segmental
hyalinizing vasculitis of the dermis. Arch Dermatol 1967; 96:489.
Alegre VA, Gastineau DA, Winkelmann RK. Skin lesions
associated with circulating lupus anticoagulant. Br J Dermatol 1989; 120:419.
Grob JJ, Bonerandi JJ. Thrombotic skin disease as a
marker of the anticardiolipin syndrome. Livedo vasculitis and distal gangrene
associated with abnormal serum antiphospholipid activity. J Am Acad Dermatol
1989; 20:1063.
Vasudevan B, Neema S, Verma R. Livedoid vasculopathy:
A review of pathogenesis and principles of management. Indian J Dermatol
Venereol Leprol 2016; 82:478.
Allenby F, Boardman L, Pflug JJ, Calnan JS. Effects of
external pneumatic intermittent compression on fibrinolysis in man. Lancet
1973; 2:1412.
Micieli R, Alavi A. Treatment for Livedoid
Vasculopathy: A Systematic Review. JAMA Dermatol 2018; 154:193.
Yang LJ, Chan HL, Chen SY, et al. Atrophie blanche. A
clinicopathological study of 27 patients. Changgeng Yi Xue Za Zhi 1991; 14:237.
El Khoury J, Taher A, Kurban M, et al. Livedoid
vasculopathy associated with sickle cell trait: significant improvement on
aspirin treatment. Int Wound J 2012; 9:344.
Maessen-Visch MB, Koedam MI, Hamulyák K, Neumann HA.
Atrophie blanche. Int J Dermatol 1999; 38:161.
Drucker CR, Duncan WC. Antiplatelet therapy in
atrophie blanche and livedo vasculitis. J Am Acad Dermatol 1982; 7:359.
Sauer GC. Pentoxifylline (Trental) therapy for the
vasculitis of atrophie blanche. Arch Dermatol 1986; 122:380.
Sams WM Jr. Livedo vasculitis. Therapy with
pentoxifylline. Arch Dermatol 1988; 124:684.
Gardette E, Moguelet P, Bouaziz JD, et al. Livedoid
Vasculopathy: A French Observational Study Including Therapeutic Options. Acta
Derm Venereol 2018; 98:842.
Weishaupt C, Strölin A, Kahle B, et al.
Characteristics, risk factors and treatment reality in livedoid vasculopathy -
a multicentre analysis. J Eur Acad Dermatol Venereol 2019; 33:1784.
Davis MD, Wysokinski WE. Ulcerations caused by
livedoid vasculopathy associated with a prothrombotic state: Response to
warfarin. J Am Acad Dermatol 2008; 58:512.
Kavala M, Kocaturk E, Zindanci I, et al. A case of
livedoid vasculopathy associated with factor V Leiden mutation: successful
treatment with oral warfarin. J Dermatolog Treat 2008; 19:121.
Browning CE, Callen JP. Warfarin therapy for livedoid
vasculopathy associated with cryofibrinogenemia and hyperhomocysteinemia. Arch
Dermatol 2006; 142:75.
Hairston BR, Davis MD, Gibson LE, Drage LA. Treatment
of livedoid vasculopathy with low-molecular-weight heparin: report of 2 cases.
Arch Dermatol 2003; 139:987.
Abou Rahal J, Ishak RS, Otrock ZK, et al. Livedoid
vasculopathy in a patient with lupus anticoagulant and MTHFR mutation:
treatment with low-molecular-weight heparin. J Thromb Thrombolysis 2012;
34:541.
Weishaupt C, Strölin A, Kahle B, et al.
Anticoagulation with rivaroxaban for livedoid vasculopathy (RILIVA): a
multicentre, single-arm, open-label, phase 2a, proof-of-concept trial. Lancet
Haematol 2016; 3:e72.
Lee JS, Cho S. Livedoid vasculopathy in Koreans:
clinical features and response to rivaroxaban treatment. J Eur Acad Dermatol
Venereol 2020; 34:e176.
Evans JM, Jensen JD, Sami N. Successful treatment of
livedoid vasculopathy with rivaroxaban. JAAD Case Rep 2015; 1:340.
Yamaguchi Y, Nakazato S, Izumi K, et al. Rapid
remission of severe pain from livedoid vasculopathy by apixaban. J Eur Acad
Dermatol Venereol 2017; 31:e45.
Furukawa F, Mizawa M, Makino T, Shimizu T. Efficacy of
new low-dose oral anticoagulants in recalcitrant livedoid vasculopathy. BMJ
Case Rep 2017; 2017.
Sawada T, Suehiro M. Dabigatran in the management of
livedoid vasculopathy. Clin Exp Dermatol 2017; 42:237.
Antunes J, Filipe P, André M, et al. Livedoid
vasculopathy associated with plasminogen activator inhibitor-1 promoter
homozygosity (4G/4G) and prothrombin G20210A heterozygosity: response to t-PA
therapy. Acta Derm Venereol 2010; 90:91.
Klein KL, Pittelkow MR. Tissue plasminogen activator
for treatment of livedoid vasculitis. Mayo Clin Proc 1992; 67:923.
Johnson DW, Hawley CM, Strutton G, Gibbs HH. Dramatic
response of livedoid vasculitis to tissue plasminogen activator (tPA). Aust N Z
J Med 1995; 25:370.
Kerk N, Drabik A, Luger TA, et al. Rivaroxaban
prevents painful cutaneous infarctions in livedoid vasculopathy. Br J Dermatol
2013; 168:898.
Juan WH, Chan YS, Lee JC, et al. Livedoid
vasculopathy: long-term follow-up results following hyperbaric oxygen therapy.
Br J Dermatol 2006; 154:251.
Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed
intravenous immunoglobulin therapy in livedoid vasculitis: an open trial
evaluating 9 consecutive patients. J Am Acad Dermatol 2004; 51:574.
Purcell SM, Hayes TJ. Nifedipine treatment of
idiopathic atrophie blanche. J Am Acad Dermatol 1986; 14:851.
Mushtaq S, Singh S, Mudugal R. Topical becaplermin gel
is an effective adjuvant for long-standing ulcers of livedoid vasculopathy
recalcitrant to anticoagulant therapy. Clin Exp Dermatol 2019; 44:681.
Lee JH, Choi HJ, Kim SM, et al. Livedoid vasculitis
responding to PUVA therapy. Int J Dermatol 2001; 40:153.
Tuchinda C, Leenutaphong V, Sudtim S, Lim HW.
Refractory livedoid vasculitis responding to PUVA: a report of four cases.
Photodermatol Photoimmunol Photomed 2005; 21:154.
Bisalbutra P, Kullavanijaya P. Sulfasalazine in
atrophie blanche. J Am Acad Dermatol 1993; 28:275.
Gupta AK, Goldfarb MT, Voorhees JJ. The use of
sulfasalazine in atrophie blanche. Int J Dermatol 1990; 29:663.
Chow M, Swift R, Sutton A, Wysong A. Endovenous Laser
Ablation Treatment for Lower Extremity Ulcers Associated With Livedoid
Vasculopathy. Dermatol Surg 2020; 46:853.
tion or impaired fibrinolysis that results in
occlusion of dermal blood vessels with fibrin thrombi [2].





