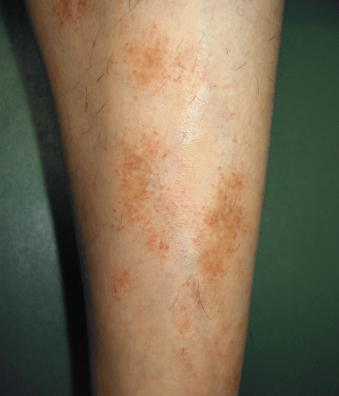
Presentamos un caso
de dermatitis purpúrica pigmentaria, más probablemente una enfermedad de
Schamberg en una paciente de 66 años con
estas lesiones en ambas piernas de varios meses de evolución.




Las lesiones no
generan prurito ni dolor y comprometen sólo miembros inferiores, no desaparecen
a la digitopresión es decir que se trata de lesiones purpúricas de aspecto en "pimienta de Cayena". La paciente no tiene antecedentes patológicos.

Gentileza
de:
Dra.
Sylvia Amórtegui de Bogotá Colombia.
DERMATOSIS
PURPÚRICAS PIGMENTADAS (CAPILARITIS)
INTRODUCCIÓN
Las
dermatosis purpúricas pigmentadas (DPP), también conocidas como capilaritis,
púrpura simple y púrpura inflamatoria sin vasculitis, son un grupo de
erupciones cutáneas crónicas benignas caracterizadas por la presencia de
petequias, púrpura y aumento de la pigmentación de la piel. Los PPD ocurren con
mayor frecuencia en las extremidades inferiores y pueden ser asintomáticos o
pruriginosos.
La
familiaridad con los hallazgos clínicos y patológicos de los DPP es útil para
distinguirlos de la vasculitis cutánea y otros trastornos que pueden
presentarse con características clínicas similares. Los hallazgos patológicos
clásicos de los DPP incluyen un infiltrado inflamatorio de células
mononucleares perivasculares, extravasación de eritrocitos y depósito de
hemosiderina.
EPIDEMIOLOGÍA
Los
datos son limitados sobre la epidemiología de las dermatosis purpúricas
pigmentadas (DPP). En general, estos trastornos se consideran poco frecuentes [
1 ]. Los principales subtipos de DPP incluyen:
- Enfermedad de Schamberg (la forma más común de DPP [ 2,3 ], también conocida como púrpura pigmentaria progresiva)
- Púrpura anular telangiectoide (también conocida como enfermedad de Majocchi)
- Dermatitis liquenoide purpúrica pigmentada de Gougerot y Blum
- Liquen aureus
- Púrpura de tipo eccematide de Doucas y Kapetanakis
Raras,
los subtipos adicionales de DPP incluyen púrpura con picazón, capilaritis
lineal unilateral y púrpura pigmentada granulomatosa. Algunos autores han
considerado la púrpura eccematídica de Doucas y Kapetanakis y la púrpura con
picazón como una sola entidad [ 4,5 ].
Aunque
las DPP puede desarrollarse a cualquier edad, se han observado predilecciones
por la edad en algunos subtipos [ 2,4,6-8 ]:
Enfermedad
de Schamberg - adultos de mediana edad
Púrpura
anular telangiectoide - adolescentes y adultos jóvenes
Dermatitis
liquenoide purpúrica pigmentada de Gougerot y Blum - adultos
Liquen
aureus - adultos jóvenes
Púrpura de tipo eccematide de Doucas y Kapetanakis -
adultos de mediana edad
Púrpura
pruriginos - adultos de mediana edad
Capilarritis
lineal unilateral - adultos jóvenes
Púrpura
pigmentaria granulomatosa - adultos
Los
niños no están excluidos del desarrollo de DPP [ 2,9 ]. La enfermedad de Schamberg
es la causa más común de petequias en niños [ 2 , 3, 9 ], y se ha documentado
en un niño de apenas un año [ 10 ]. Además, hay múltiples informes publicados
de liquen aureus en niños [ 11-19 ].
Los
datos son insuficientes para confirmar si existen predilecciones de género
entre los DPP, y dos de las series más grandes han encontrado resultados
contradictorios sobre si la enfermedad de Schamberg es más común en hombres o
mujeres [ 5,7 ]. Aunque tampoco hay datos suficientes para establecer conclusiones
sobre predilecciones raciales o étnicas para los DPP, se ha observado que los
pacientes de ascendencia del este de Asia constituyen la mayoría de los casos
reportados de púrpura pigmentada granulomatosa [ 20-25 ].
ETIOLOGÍA
Y PATOGENIA
Se han
propuesto múltiples factores como contribuyentes potenciales al desarrollo de
dermatosis purpúricas pigmentadas (PPD), aunque ninguno ha sido probado
definitivamente. Estos incluyen [ 2,4,21 ]:
- Hipertensión venosa
- Dependencia gravitacional
- Ejercicio aerobico
- Infección localizada [ 26 ]
- Alcohol [ 27 ]Ingestión química
- Alergenos de contacto ( tabla 1 ) [ 28-38 ]
- Drogas ( tabla 2 ) [ 39-51 ]
- Enfermedades sistémicas

Tabla 1. Alergenos de contacto relacionados con dermatitis purpúricas pigmentadas


Tabla 2. Medicamentos asociados a dermatitis purpúricas pigmentadas.
En los
DPP relacionados con la exposición a medicamentos, se han informado tiempos de
inicio que van desde una semana hasta años después del inicio del tratamiento
farmacológico [ 39-49 ].
Se han
propuesto asociaciones entre DPP y múltiples enfermedades sistémicas [ 5 ]. Los
ejemplos de las asociaciones más comúnmente citadas incluyen hiperlipidemia (PPD
granulomatosa) [ 21,25,52,53 ] y hepatitis viral [ 54-57 ]. Sin embargo, la
relación entre DPP y estos trastornos es incierta. Un estudio de casos y
controles de 60 pacientes con PPD y 230 controles no encontró diferencias
significativas en la prevalencia de hepatitis B o C entre los pacientes con PPD
[ 58 ]. Las PPD no son manifestaciones de anormalidades plaquetarias o
trastornos hemorrágicos.
Algunos
autores han considerado la DPP como una forma de discrasia linfoide cutánea de
células T basada en informes poco frecuentes de DPP que progresa a micosis
fungoide, similitudes en las características clínicas e histológicas de DPP y
micosis fungoide, y la detección frecuente de monoclonalidad de células T en
lesiones de DPP [ 59 ] Se necesitan más estudios para aclarar la relación entre
micosis fungoide y DPP.
Hay una
escasez de datos sobre los mecanismos patogénicos que conducen a los hallazgos
clínicos de DPP, y por lo tanto, los mecanismos a través de los cuales se
desarrolla DPP son poco conocidos. A continuación se revisan tres teorías que
han surgido en base a la interpretación de los hallazgos histológicos e
inmunohistoquímicos en DPP:
Anormalidades
vasculares: la extravasación de eritrocitos en la dermis es un rasgo patológico
característico de la DPP. Se ha postulado un estado de fragilidad capilar para
contribuir a la ruptura capilar y la extravasación de eritrocitos, consistente
con lo observado en la DPP [ 2 ]. El aumento de la presión en la
microvasculatura cutánea secundaria a estasis venosa también se ha propuesto
como un factor contribuyente [ 60 ].
Inmunidad
mediada por células: un infiltrado de células inflamatorias mononucleares de
grado variable se encuentra típicamente en muestras patológicas de PPD, y es
concebible que una respuesta inmune mediada por células perivasculares pueda
contribuir al daño capilar y la fuga de eritrocitos [ 61 ]. La naturaleza del
infiltrado inflamatorio en la enfermedad de Schamberg se investigó en un
estudio de muestras de tejido de cinco adultos con la afección [ 62 ]. El
infiltrado estaba compuesto principalmente por linfocitos CD4 + y células
dendríticas CD1a + . Además, se detectó una regulación positiva de las
moléculas de adhesión que podrían facilitar el tráfico de células inflamatorias
en los linfocitos infiltrantes (LFA-1 e ICAM-1) y las células endoteliales
(ICAM-1 y ELAM-1).
Inmunidad
humoral : se ha sugerido un papel potencial para la enfermedad del complejo
inmunitario en la PPD en base a informes que documentan hallazgos de
inmunofluorescencia directa de depósitos vasculares de C3, C1q, IgM o IgA
dentro de la piel lesionada [ 5,63,64 ].
Como
consecuencia de la investigación limitada, el papel de cada uno de estos
factores en el desarrollo de PPD sigue siendo incierto. Se necesitan estudios
adicionales para dilucidar la fisiopatología de este trastorno.
CARACTERÍSTICAS
CLÍNICAS
Las
características clínicas clave de todas las dermatosis purpúricas pigmentadas
(DPP) son petequias o púrpura, que reflejan la extravasación de eritrocitos, y
parches pigmentados de color amarillo a marrón, que son el resultado del
depósito de hemosiderina dentro de la dermis. Los sitios más comunes para DPP
son las extremidades inferiores, aunque las lesiones también pueden
desarrollarse en otras áreas. Con poca frecuencia, los DPP se presentan como
erupciones generalizadas. Las palmas, las plantas de los pies, los genitales y
la mucosa generalmente están respetadas.
Los
hallazgos clínicos que distinguen los subtipos de DPP se revisan a
continuación.
ENFERMEDAD
DE SCHAMBERG (PÚRPURA PIGMENTARIA PROGRESIVA)
Descrita
por primera vez por Jay Frank Schamberg en 1901 [ 65 ], la enfermedad de
Schamberg se presenta con parches purpúreos discretos, que no desaparecen con
la vitropresión, de color marrón rojizo que generalmente tienen unos pocos
milímetros a varios milímetros de diámetro, aunque algunos parches pueden
alcanzar varios centímetros de ancho ( imágenes 5,6,7 ). Una inspección
minuciosa de los parches revela petequias puntiformes no palpables que
Schamberg describió como "puntiformes a puntas rojizas del tamaño de una
cabeza de alfiler, muy parecidas a los granos de pimienta de cayena" [ 65]
El sitio más común para la participación son las extremidades inferiores
bilaterales (debajo de las rodillas). Los muslos y las nalgas también están
frecuentemente involucrados. Con menos frecuencia, la enfermedad de Schamberg
se presenta con extremidad superior o afectación generalizada. Aunque la
enfermedad de Schamberg suele ser asintomática, los pacientes ocasionales
experimentan prurito leve.
Figura
5. Enfermedad de Schamberg
Las
máculas y parches marrones con rojo-marrón (similar a la pimienta de cayena),
máculas puntiformes están presentes en este paciente con enfermedad de
Schamberg, una variante de la dermatosis purpúrica pigmentada.

Figura
6. Enfermedad de Schamberg
Múltiples
petequias y púrpura en las extremidades inferiores en la enfermedad de
Schamberg (púrpura pigmentaria progresiva).

Figura
7. Enfermedad de Schamberg
La
compresión con un portaobjetos de vidrio revela la naturaleza no blanqueable de
las petequias y la púrpura en la enfermedad de Schamberg (púrpura pigmentaria
progresiva), un tipo de dermatosis purpúrica pigmentada.
El curso
de la afección es típicamente crónica, con numerosas exacerbaciones y remisiones.
La resolución espontánea también puede ocurrir [ 5,9,66-70 ]. En una serie
retrospectiva que incluyó al menos tres años de datos de seguimiento de cada
uno de los 45 pacientes con enfermedad de Schamberg, el 62 por ciento se aclaró
o mejoró y el 38 por ciento se mantuvo estable o experimentó un empeoramiento
de los síntomas [ 5 ].
PURPURA
ANULAR TELANGIECTODE (ENFERMEDAD DE MAJOCCHI)
Inicialmente
descrito por Domenico Majocchi en 1896 [ 71 ], purpura annularis telangiectodes
se presenta con parches no blanqueables, anulares, de 2 a 20 cm, simétricos,
purpúricos, telangiectásicos ( imágene 8 y 9 ). Se informó enfermedad
unilateral en un paciente [ 72 ]. Parches lineales o arciformes también pueden
estar presentes. Ocasionalmente, la atrofia es evidente en el centro de las
lesiones [ 73 ].

Figura
8. Púrpura anular telangiectoide (enfermedad de Majocchi)
Hay
parches purpúricos anulares en la pierna.

Figura
9. Púrpura anular telangiectoide (enfermedad de Majocchi)
Gran
parche purpúrico en la extremidad inferior con una configuración anular.
Al igual
que con la enfermedad de Schamberg, el sitio más común para involucrarse son
las extremidades inferiores [ 74 ]. Puede ocurrir extensión a las extremidades
superiores y al tronco.
Las
lesiones cutáneas de la púrpura anular telangiectodes generalmente son
asintomáticas. Ocasionalmente, el prurito leve está presente [ 73,75 ]. El
curso es crónico y se caracteriza por recaídas y remisiones [ 73 ]. Las
lesiones individuales suelen durar varios meses.
DERMATITIS
LIQUENOIDE PURPÚRICA PIGMENTADA DE GOUGEROT Y BLUM
Los pacientes con dermatitis liquenoide purpúrica
pigmentada de Gougerot y Blum desarrollan pequeñas pápulas liquenoides purpúricas
poligonales o redondas que se unen para formar placas de color marrón rojizo a
violáceo de uno a varios centímetros de diámetro ( imagen 10 ) [ 76 ]. La
apariencia de las placas puede parecerse al sarcoma de Kaposi [ 77 ]. Al igual
que otros subtipos de DPP, esta erupción generalmente se encuentra en las
extremidades inferiores. En raras ocasiones, hay afectación de las extremidades
superiores o el tronco. El curso de la dermatitis liquenoide purpúrica
pigmentada de Gougerot y Blum tiende a ser crónica, aunque puede producirse una
remisión espontánea [ 4 ].

Figura
10. Dermatosis liquenoide purpúrica pigmentada de Gougerot y Blum.
Pápulas
violáceas y pequeñas placas, así como máculas de color amarillo-marrón en las
extremidades inferiores.
LIQUEN
AUREUS
El liquen aureus (originalmente denominado
"liquen purpúrico") se presenta con la aparición aguda de pápulas
liquenoides distintivas, de color amarillo óxido o dorado o parches y placas
circunscritas ( figura 11 ) [ 78 ]. El término "liquen aureus"
enfatiza el clásico color marrón dorado de la erupción [ 79 ]. Cabe destacar
que el liquen aureus también puede presentarse con lesiones cobre anaranjado o violáceas.

Figura
11. Liquen aureus.
El
liquen aureus ocurre con mayor frecuencia de manera unilateral en las
extremidades inferiores; La afectación bilateral es menos frecuente [ 11,15,80
]. Otras distribuciones reportadas incluyen la extremidad superior y la
afectación del tronco; arreglos segmentarios, dermatomales y blascoides; e
implicación que sigue el curso de las venas superficiales o profundas [ 80-85
]. Las ubicaciones y distribuciones aberrantes pueden ocurrir más comúnmente en
niños [ 11 ].
El
liquen aureus generalmente es asintomático. Ocasionalmente, los pacientes
experimentan prurito intenso.
El
liquen aureus tiende a persistir por varios años y, en algunos casos, es lentamente
progresivo [ 80,86 ]. En un estudio retrospectivo de 42 pacientes con liquen
aureus (edad de inicio de 3 a 70 años), las lesiones tendieron a persistir
durante el período de seguimiento de 3 meses a 19 años [ 80 ]. La resolución
completa se informó solo para dos pacientes. En contraste, la resolución
espontánea se observó con mayor frecuencia en una serie de ocho niños con
liquen aureus; cinco experimentaron resolución en dos a seis años (media 3.4
años), dos habían reducido pero continuaron las lesiones más allá de cuatro a
cinco años, y uno tenía lesiones persistentes a los tres años [ 11 ].
PÚRPURA
SIMILAR ECZEMATIDE DE DOUCAS Y KAPETANAKIS
La
púrpura eczematid de Doucas y
Kapetanakis se distingue de otras formas de DPP por la presencia concomitante
de características eccematosas. La erupción generalmente comienza con la
aparición insidiosa de grupos de máculas rojas puntiagudas no blanqueables que
evolucionan a un color rojo oscuro o amarillo-marrón durante unas pocas semanas
[ 6 ]. La erupción generalmente comienza en las extremidades inferiores y puede
extenderse a las extremidades superiores o al tronco. La cara, el cuello, las
palmas y las plantas generalmente están libres.
El
eritema y la escala leve se desarrollan dentro de las áreas involucradas, lo
que resulta en una apariencia que imita el eccema ( imagen 12 y 13 ). El
prurito es común. Puede haber liquenificación leve secundaria al roce y al
rascado.

Figura
12. Púrpura eccematide de Doucas y Kapetanakis
Parches
eccematosos purpúricos con escamas leves en las extremidades.

Figura
13. Púrpura eccematide de Doucas y Kapetanakis
Purpúrico,
parches eritematosos con escamas.
La
púrpura de tipo eccematida de Doucas y Kapetanakis sigue un curso fluctuante.
La erupción generalmente se resuelve espontáneamente dentro de varios meses a
dos años [ 6 ].
VARIANTES
RARAS
Variantes
raras ocasiones reportados de DPP incluyen la púrpura pruriginosa, la capilaritis
lineal unilateral, y la púrpura pigmentada granulomatosa:
PÚRPURA
PRURIGINOSA (ITCHING PURPURA )
La
púrpura pruriginosa (también conocida como angiodermatitis pruriginosa
diseminada) se caracteriza por la aparición aguda de máculas de color marrón
anaranjado a purpúrico en las extremidades inferiores. Las lesiones a menudo se
generalizan posteriormente. A diferencia de la mayoría de los otros tipos de
DPP, la afección se asocia con prurito severo [ 87,88 ]. La púrpura que pica
tiene un curso crónico, aunque ocurren remisiones espontáneas.

Figura
14. Púrpura pruriginosa (itching purpura)
CAPILARITIS
LINEAL UNILATERAL
La
capilaritis lineal unilateral (también conocida como púrpura segmentada
pigmentada y capilaropatía cuadrática) describe el desarrollo de la púrpura
pigmentada en una distribución lineal, dermatómica o segmentaria ( imagen 15 )
[ 89-93 ]. La condición se ha informado principalmente en adultos jóvenes y
niños en las extremidades inferiores o superiores [ 89,90 ]. El pronóstico
parece ser bueno; En una serie de cuatro hombres con lesiones en las
extremidades inferiores, en todos los pacientes se produjo una regresión
significativa de las lesiones cutáneas en tres años [ 89 ].

Figura
15. Capilaritis lineal unilateral.
Parches
marrones y purpúricos en una distribución lineal en la extremidad inferior.
PÚRPURA
PIGMENTADA GRANULOMATOSA: se han notificado aproximadamente 20 casos de púrpura
pigmentada granulomatosa en la literatura [ 20-25,52,53,94-96 ]. La afección
generalmente se presenta con máculas o pápulas purpúricas eritematosas a
marrones múltiples y se distingue de otros tipos de DPP por la detección de un
infiltrado granulomatoso en la histopatología ( imagen 16, 17 ). Los sitios de
participación más comunes son el dorso de los pies y las piernas. Con menos
frecuencia, muñecas y manos han sido afectadas. También se ha informado de una
distribución de blaschkoides [ 97 ].

Figura
16.Púrpura pigmentada granulomatosa
Pápulas
purpúricas marrones en la parte inferior de las piernas y el dorso de los pies.

Figura
17. Púrpura pigmentada granulomatosa
Vista
del infiltrado granulomatoso en la dermis superficial con extravasación de
glóbulos rojos y siderófagos.
Muchos
de los casos reportados han ocurrido en pacientes que también tenían
hiperlipidemia, lo que llevó a algunos autores a cuestionarse si existe una
relación entre estos trastornos [ 25,52,53,94,95 ]. Las lesiones cutáneas
pueden persistir durante años [ 52 ].
HISTOPATOLOGIA
Los
subtipos de dermatosis purpúrica pigmentada (DPP) comparten varias
características histopatológicas, que incluyen [ 5,59,98 ]:
- Un infiltrado inflamatorio perivascular superficial (principalmente linfocitos CD4 + con ocasionalmente células dendríticas y macrófagos)
- Proliferación de células endoteliales y edema; estrechamiento de lúmenes de vasos
- Extravasación de eritrocitos en la dermis papilar
- Grado variable de deposición de hemosiderina en macrófagos en la dermis (particularmente en lesiones maduras)
- Exocitosis linfocitaria
- Degeneración vacuolar leve variable en la capa basal
Las
tinciones histoquímicas que tiñen el hierro, como la tinción de Perls o Fontana
Masson, son útiles para resaltar el depósito de hemosiderina en la dermis
superficial. Es de destacar que las características de la vasculitis
leucocitoclástica (p. Ej., Leucocitoclasia y necrosis fibrinoide de las paredes
de los vasos) están ausentes.
LABORATORIO
No hay
anomalías de laboratorio asociadas con dermatosis purpúricas pigmentadas (DPP).
Se espera que las pruebas de laboratorio que evalúan anormalidades
plaquetarias, coagulopatías o reactivos de fase aguda sean normales.
DIAGNÓSTICO
La
presencia de máculas y parches purpúricos discretos o circunscritos, no
palpables (particularmente en la extremidad inferior) con petequias y
pigmentación amarillo-marrón sugiere la posibilidad de dermatosis purpúricas
pigmentadas (DPP), particularmente la enfermedad de Schamberg, la forma clásica
de DPP. Las lesiones palpables que exhiben características similares sugieren
variantes de DPP que a menudo se presentan con pápulas o placas, como liquen
aureus, púrpura eccematídice de Doucas y Kapetanakis y púrpura pigmentada
granulomatosa. El diagnóstico generalmente se realiza mediante inspección
clínica y el reconocimiento de las características morfológicas clásicas.
La
dermatoscopia puede ser un complemento útil para la inspección clínica a simple
vista. Una revisión de 32 pacientes con DPP sugiere que los hallazgos
dermatoscópicos en lesiones de púrpura pigmentada pueden incluir pigmentación
rojo cobrizo en el fondo; una red marrón; vasos lineales; redondo a ovalado,
puntos rojos, glóbulos y parches; glóbulos y puntos marrones; línea lineal
marrón; y aberturas foliculares [ 100 ].
Una
biopsia de piel es útil cuando el diagnóstico sigue siendo incierto después del
examen clínico. Las biopsias pueden ayudar a distinguir DPP de los trastornos
en el diagnóstico diferencial, como la vasculitis cutánea.
DIAGNÓSTICO
DIFERENCIAL
El
diagnóstico diferencial de las dermatosis purpúricas pigmentadas (DPP) consiste
en otras lesiones cutáneas que pueden presentarse con petequias, púrpura,
características eccematosas o hiperpigmentación localizada relacionada con el
depósito de hemosiderina. Una evaluación clínica cuidadosa y una biopsia
(cuando sea necesario) pueden ayudar a distinguir DPP de otros trastornos. La
dermatitis por estasis, la vasculitis cutánea, la púrpura traumática y la
micosis fungoide se encuentran entre los trastornos que pueden confundirse con DPP:
DERMATITIS
POR ESTASIS
La
dermatitis por estasis comparte características clínicas y patológicas con DPP.
La dermatitis por estasis generalmente se presenta en las extremidades
inferiores con inicio insidioso de parches eccematosos que progresan a parches
irregulares de color rojo-marrón o musculoso ( imagen 18,19 ). Varices
asociadas y edema de la pierna a menudo están presentes. El tobillo medial es
el sitio más común de afectación inicial. Al igual que las DPP, los hallazgos
patológicos de la dermatitis por estasis incluyen espongiosis epidérmica, un
infiltrado linfocítico perivascular, capilares dérmicos dilatados con
engrosamiento intimal y hemosiderina dentro de los macrófagos dérmicos. Los
médicos deben tener en cuenta que la dermatitis por estasis y la DPP pueden
coexistir.

Figura
18. Dermatitis de estasis temprana.
Este
paciente tiene el primer signo de dermatitis por estasis: eritema, descamación
e hiperpigmentación leve en la región supramalleolar medial.

Figura
19. Petequias en dermatitis por estasis.
Numerosas
petequias están presentes en la parte inferior de la pierna en este paciente
con dermatitis por estasis.
VASCULITIS
CUTÁNEA
A veces
las vasculitis leucocitoclástica cutánea se presentan al inicio agudo con púrpura palpable , a menudo
afecta principalmente a las extremidades inferiores y otras áreas dependientes
del cuerpo. También pueden estar presentes ampollas hemorrágicas y pigmentación
marrón consistente con el depósito de hemosiderina en la piel ( imagen
20,21,22,23 ). La naturaleza palpable de este trastorno es útil para
distinguirlo de la enfermedad de Schamberg y la púrpura anular telangiectoide,
que se presentan con petequias y / o púrpura no palpables . Una biopsia de piel
puede distinguir la vasculitis de la DPP; La destrucción de los vasos, la
leucocitoclasia y la necrosis fibrinoidea característica de la vasculitis no se
encuentran en la DPP.

Figura
20. Púrpura palpable envasculitis cutánea
Lesiones
cutáneas purpúricas palpables, no blanqueantes, que se encuentran en la
extremidad inferior de un paciente con vasculitis de vasos pequeños que afecta
la piel.

Figura
21. Púrpura palpable en vasculitis cutánea
La
púrpura palpable está presente en las piernas de este paciente con vasculitis
cutánea de vasos pequeños.

Figura
22. Púrpura palpable en vasculitis cutánea
La
púrpura palpable está presente en este paciente con vasculitis cutánea de vasos
pequeños.

Figura
23. Vasculitis cutánea de vasos pequeños ampollar
Las
vesículas que recubren las máculas purpúricas están presentes en la parte
inferior de la pierna.
PÚRPURA
TRAUMÁTICA
La
lesión de la piel puede provocar el desarrollo de parches purpúricos. La
púrpura traumática es mucho menos persistente que los DPP, por lo general
evoluciona y se resuelve en el transcurso de unas pocas semanas.
MICOSIS
FUNGOIDE
Las PPD son veces difíciles de distinguir de la micosis
fungoide. Con poca frecuencia, la púrpura es una manifestación inicial de
micosis fungoide, y los dos trastornos pueden tener características
histológicas superpuestas (p. Ej., Infiltrado liquenoide, linfocitos que
invaden la epidermis). En los casos en que las características clínicas
sugieren DPP, pero el examen patológico plantea la cuestión de la micosis
fungoide, distinguir entre las condiciones puede ser un desafío. La detección
de grandes grupos intraepidérmicos de linfocitos o de muchos linfocitos en la
capa espinosa de la epidermis favorece el diagnóstico de micosis fungoide [ 101
].
ESCORBUTO
Las
lesiones purpúricas del escorbuto pueden confundirse con las petequias
dispersas y la púrpura de la enfermedad de Schamberg ( imagen 10A-B ). La
detección de púrpura perifolicular, pelos en forma de sacacorchos, encías
sangrantes y un historial dietético de ingesta reducida de vitamina C respaldan
un diagnóstico de escorbuto.

Figura
24. Escorbuto
Las
máculas purpúricas perifoliculares están presentes en este paciente con
escorbuto.

Figura
25. Anomalías gingivales en el escorbuto
La
hinchazón gingival y el color oscuro justo por encima de dos de los dientes
indican hemorragia en las encías de este paciente con mala dentición. Las
anormalidades gingivales del escorbuto ocurren solo en presencia de dientes,
que presumiblemente proporcionan portales de entrada para los microbios en las
encías. Una hipótesis sugiere que la deficiencia de vitamina C perjudica la
muerte de bacterias mediada por neutrófilos, lo que lleva a la gingivitis
crónica, que luego se complica por el sangrado de los vasos frágiles
característicos del escorbuto.
DIAGNÓSTICOS
DIFERENCIALES DE LA DERMATITIS ECCEMATOSA
DE DOUCAS Y KAPETANAKIS
Estas
entidades incluyen dermatitis atópica, eccema numular y dermatitis alérgica de
contacto ( imagen 26,27,28,29 ). Estos trastornos pueden distinguirse clínicamente
de este subtipo de DPP por la ausencia de petequias, púrpura y la pigmentación
amarillo-marrón característica de los DPP. Además, los hallazgos patológicos de
la dermatitis atópica y la dermatitis alérgica de contacto no incluyen la
extravasación de eritrocitos y el depósito de hemosiderina.

Figura 26.
Dermatitis atópica en adultos
Dermatitis
atópica crónica con liquenificación (engrosamiento de la piel y mejora de las
marcas de la piel) de las flexiones de la rodilla en una mujer de 22 años.

Figura
27. Dermatitis atópica crónica del adulto
Placa
liquenificada e hiperpigmentada en la flexión del codo de una mujer de 35 años
con dermatitis atópica.

Figura
28. Eccema numular.
Los
parches y placas "en forma de moneda" se encuentran en las piernas.

Figura
29. Dermatitis alérgica de contacto.
Dermatitis
alérgica de contacto de la parte superior del pie por zapatos de cuero (alergia
al cromato).
SARCOMA
DE KAPOSI
El
sarcoma de Kaposi puede imitar la presentación de dermatitis liquenoide
purpúrica pigmentada de Gougerot y Blum. Las lesiones cutáneas del sarcoma de
Kaposi aparecen como máculas, pápulas, placas o nódulos violáceos, más
comúnmente en las extremidades inferiores, la cabeza, el cuello y la mucosa
oral ( imagen 30 ). Los últimos tres sitios generalmente están libres de DPP.
El sarcoma de Kaposi se distingue fácilmente del DPP a través de hallazgos
patológicos de proliferación vascular, células fusiformes dérmicas y hendiduras
vasculares.

Figura
30. Sarcoma de Kaposi.
TRATAMIENTO
Las
dermatosis purpúricas pigmentadas (DPP) son afecciones benignas que a menudo
son asintomáticas. Por lo tanto, el tratamiento no siempre es necesario y la
tranquilidad es un curso de tratamiento aceptable para la mayoría de los
pacientes. El tratamiento generalmente se considera para pacientes con síntomas
asociados y para pacientes que están angustiados por la apariencia estética de
las lesiones cutáneas.
Debido a
que la mayoría de los datos sobre la eficacia de los tratamientos se derivan de
informes de casos y series de casos pequeños y es posible la resolución
espontánea de DPP, el mejor enfoque para el tratamiento sigue siendo incierto.
En nuestra experiencia, la respuesta al tratamiento es impredecible y, a
menudo, pobre. Dada la naturaleza benigna de los DPP, siempre debe considerarse
la relación riesgo-beneficio del tratamiento.
Cuando
los pacientes desean tratamiento, generalmente comenzamos con medidas para
reducir los factores de exacerbación o los corticosteroides tópicos debido a la
relativa seguridad de estas intervenciones. Reservamos otras intervenciones
para pacientes con compromiso extenso o lesiones tópicas refractarias a
corticosteroides que también tienen un fuerte deseo de tratamiento adicional.
Intervenciones
de primera línea : nuestras terapias de primera línea para los
DPP son intervenciones no farmacológicas y corticosteroides tópicos.
LAS
INTERVENCIONES NO FARMACOLÓGICAS - las intervenciones no farmacológicas para DPP están dirigidos a eliminar o reducir los
posibles factores contribuyentes. En pacientes en los que el desarrollo de un DPP parece correlacionarse con la exposición a un medicamento o alérgeno de
contacto, se debe implementar la interrupción de la exposición al posible
agente incitante siempre que sea posible.
Debido a
la teoría de que el aumento de la presión vascular puede contribuir a la fuga
capilar en los DPP, muchos médicos, incluido nosotros mismos, a menudo tienen
pacientes con DPP en las extremidades inferiores que usan medias de apoyo,
particularmente si hay signos concomitantes de estasis venosa. Sin embargo, la
eficacia de esta medida para acelerar la resolución del DPP no está probada.
Los
corticosteroides tópicos - Los corticosteroides tópicos se han utilizado
para DPP en base a la evidencia histológica que DPP es un proceso inflamatorio.
Aunque algunos informes de casos y series de casos han documentado una mejora
en la DPP durante la terapia tópica con corticosteroides, la respuesta al
tratamiento es inconsistente [ 4,5,9,13,25,102-104 ]. No se han realizado
ensayos controlados aleatorios de la terapia con corticosteroides tópicos para
DPP.
Cuando
se trata con corticosteroides tópicos, generalmente prescribimos la aplicación
de un corticosteroide tópico de potencia media o alta de una a dos veces al día
en las áreas afectadas durante cuatro a seis semanas [ 1 ]. Si no se detecta mejoría dentro de este
período, descontinuamos el tratamiento tópico con corticosteroides.
La
atrofia cutánea es un posible efecto adverso del tratamiento tópico con
corticosteroides. Los efectos adversos de este tratamiento se revisan con mayor
detalle por separado.
Intervención
de segunda línea: la fototerapia es un
tratamiento bien tolerado dirigido a la piel que parece ser valioso para
algunos pacientes con DPP. Consideramos la fototerapia para pacientes que
fracasan con la terapia tópica con corticosteroides o para pacientes para
quienes la terapia tópica no es práctica debido a una participación extensa.
Los datos sobre el beneficio a largo plazo de la fototerapia después del cese
del tratamiento son limitados.
Fototerapia: los informes de casos y las pequeñas
series de casos han documentado una mejora en los PPD con fototerapia
ultravioleta B (UVB) o psoraleno más ultravioleta A (PUVA). Se requieren
múltiples sesiones de tratamiento. Las visitas repetidas requeridas para la
fototerapia son un elemento disuasorio para el tratamiento de algunos pacientes
(ver "terapia UVB (banda ancha y banda estrecha)" y "Psoraleno
más fotoquimioterapia ultravioleta A (PUVA)" ):
- UVB de banda estrecha : en un estudio prospectivo no controlado de cinco pacientes con enfermedad de Schamberg y un paciente con telangiectodos de púrpura anular, el tratamiento con UVB de banda estrecha tres veces por semana condujo a una buena respuesta (más del 75 por ciento de reducción en petequias y pigmentación) en todos los pacientes dentro de 24 a 28 sesiones de tratamiento [ 105 ]. Posteriormente, los tratamientos de mantenimiento se administraron dos veces por semana durante tres semanas seguidos de una vez por semana durante tres semanas. Los dos pacientes que recayeron durante el período de seguimiento de un año mejoraron con sesiones adicionales de tratamiento con UVB de banda estrecha.Los informes de casos y las series de casos describen a otros pacientes con PPD [ 106-109 ] (incluidos niños [ 9,109,110 ]) que parecían responder bien a la fototerapia UVB de banda estrecha. Por ejemplo, los pacientes con dermatitis liquenoide purpúrica pigmentada de Gougerot y Blum han logrado una mejoría o eliminación de la lesión durante la fototerapia UVB de banda estrecha [ 9,111 ].
La
mejora en DPP durante PUVA también se ha informado en informes de casos de
pacientes con enfermedad de Schamberg [ 113 ], liquen aureus [ 114 ] y
capilaritis lineal unilateral [ 90 ].
Los
posibles efectos adversos de la fototerapia incluyen quemaduras solares,
ampollas, prurito y envejecimiento de la piel. Se ha informado un mayor riesgo
de cáncer de piel en pacientes con psoriasis que han recibido un gran número de
tratamientos con PUVA. Los efectos secundarios de la fototerapia se revisan con
mayor detalle por separado. (Consulte "Terapia UVB (banda ancha y banda
estrecha)", sección sobre "Efectos adversos a corto y largo
plazo" y "Psoraleno más fotoquimioterapia ultravioleta A
(PUVA)", sección sobre "Efectos adversos" .
Otras
intervenciones se han utilizado varias otras terapias para
la DPP. Solo se ha evaluado la pentoxifilina en un ensayo aleatorizado.
Pentoxifilina:
la pentoxifilina puede tener efectos
beneficiosos sobre la enfermedad de Schamberg. La pentoxifilina es un fármaco
que inhibe la adhesión de los leucocitos a las células endoteliales y puede
estimular el endotelio vascular para liberar PGI 2 , que inhibe la adhesión y
agregación plaquetaria [ 85 ]. La pentoxifilina también se usa para el
tratamiento de la enfermedad venosa crónica de las extremidades inferiores.
Un
ensayo aleatorio simple ciego de 112 adultos con enfermedad de Schamberg de
moderada a grave comparó ocho semanas de tratamiento con pentoxifilina (400 mg
tres veces al día) con crema tópica de dipropionato de betametasona al 0.05%
(aplicado dos veces al día durante ocho semanas) [ 115 ]. Mientras que 34
pacientes en el grupo de pentoxifilina (61 por ciento) tuvieron una reducción
estadísticamente significativa en un puntaje de gravedad de la enfermedad
después de dos meses, esto ocurrió en solo 12 de 56 pacientes en el grupo de
corticosteroides (21 por ciento). A los cuatro meses después de la interrupción
del tratamiento, las reducciones estadísticamente significativas en la
puntuación de gravedad se mantuvo en 14 pacientes en el grupo de pentoxifilina
y 8 pacientes en el grupo de corticosteroides. Si se obtuvieron respuestas
cosméticamente significativas no está claro.
Los
informes de casos y pequeñas series de casos de pentoxifilina para la
enfermedad de Schamberg han documentado una mejoría clínica marcada en
pacientes con 300 mg por día o 400 mg tres veces al día [ 116,117 ] y el
fracaso del tratamiento en pacientes tratados con 300 a 600 mg por día [
118,119 ]. Se necesitan más estudios para confirmar el valor clínico del
medicamento para la PPD. Se informó una mejoría parcial después de la terapia
de combinación con pentoxifilina y prostaciclina (IGP 2 ) en un paciente con
liquen aureus segmentario [ 85 ].
El
tratamiento con pentoxifilina generalmente se tolera bien. Los síntomas
gastrointestinales (p. Ej., Náuseas, vómitos) son los efectos adversos más
comunes.
Otros: otras terapias que se ha informado que son
beneficiosas para DPP en un pequeño número de pacientes incluyen griseofulvina
[ 120 ], dobesilato de calcio (500 mg dos veces al día) [ 121 ], colchicina
(0.5 mg dos veces al día) [ 122,123 ], pomada de tacrolimus [ 124 ], crema de
pimecrolimus [ 73 ], terapia fotodinámica [ 125 ] y láser de colorante de pulso
[ 126,127 ]. Inmunosupresores tales como glucocorticoides sistémicos [ 106 ],
ciclosporina [ 128 ] y metotrexato [73 ] se han utilizado con éxito, pero
generalmente no están indicados dados los posibles efectos adversos de estos
agentes y los datos mínimos para respaldar su eficacia.
Se ha informado
que un bioflavonoide oral (rutósido) 50 mg dos veces al día y ácido ascórbico
(500 mg dos veces al día) mejoran la PPD [ 6.129.130 ]. En una serie de casos
retrospectivos de dos centros de 35 pacientes con enfermedad de Schamberg que
recibieron 50 mg de rutósido dos veces al día y 1000 mg de ácido ascórbico una
vez al día con una duración media del tratamiento de 8,2 meses, el 71 por
ciento experimentó un aclaramiento completo y el 20 por ciento experimentó una
mejora de más del 50 por ciento . Nueve participantes (25 por ciento) recayeron
después de la interrupción. Siete de estos pacientes respondieron a la
reiniciación de rutósido y ácido ascórbico. Aquellos con una menor duración de
la enfermedad tuvieron mejores respuestas, menor tiempo de respuesta y menor
riesgo de recurrencia [ 130 ].
FUENTE: UPTODATE JULIO 2020
Referencias
Sardana
K, Sarkar R, Sehgal VN. Dermatosis purpúricas pigmentadas: una visión general.
Int J Dermatol 2004; 43: 482.
Tristani-Firouzi
P, Meadows KP, Vanderhooft S. Erupciones purpúricas pigmentadas de la infancia:
una serie de casos y revisión de la literatura. Pediatr Dermatol 2001; 18: 299.
Torrelo
A, Requena C, Mediero IG, púrpura de Zambrano A. Schamberg en niños: una
revisión de 13 casos. J Am Acad Dermatol 2003; 48:31.
Newton
RC, Raimer SS. Erupciones purpúricas pigmentadas. Dermatol
Clin 1985; 3: 165.
Ratnam KV, Su WP, Peters MS. Púrpura simple (púrpura inflamatoria
sin vasculitis): un estudio clínico-patológico de 174 casos. J Am Acad Dermatol
1991; 25: 642.
DOUCAS
C, KAPETANAKIS J. Púrpura similar a eczematid. Dermatologica 1953; 106: 86.
RANDALL
SJ, KIERLAND RR, MONTGOMERY H. Erupciones purpúricas pigmentadas. AMA Arch Derm
Syphilol 1951; 64: 177.
Sherertz
EF. Erupciones purpúricas pigmentadas. Semin Thromb Hemost 1984; 10: 190.
Coulombe
J, Jean SE, Hatami A, et al. Dermatosis purpúrica pigmentada: caracterización
clinicopatológica en una serie pediátrica. Pediatr Dermatol 2015; 32: 358.
Zvulunov
A, Avinoach I, Hatskelzon L, Halevy S. Dermatosis purpúrica pigmentada (púrpura
de Schamberg) en un bebé. Dermatol Online J 1999; 5: 2.
Gelmetti
C, Cerri D, Grimalt R. Lichen aureus en la infancia. Pediatr Dermatol 1991; 8:
280.
Kahana
M, Levy A, Schewach-Millet M, y col. Liquen aureus que ocurre en la infancia.
Int J Dermatol 1985; 24: 666.
Moche J,
Glassman S, Modi D, Grayson W. Liquen aureus segmentario: un informe de dos
casos tratados con aceponato de metilprednisolona. Australas J Dermatol 2011;
52: e15.
Böhm M,
Bonsmann G, Luger TA. Resolución de liquen aureus en un niño de 10 años después
de pimecrolimus tópico. Br J Dermatol 2004; 151: 519.
Fink-Puches
R, Wolf P, Kerl H, Cerroni L. Lichen aureus: características
clinicopatológicas, historia natural y relación con micosis fungoide. Arch
Dermatol 2008; 144: 1169.
Kim MJ, Kim BY, Park KC, Youn SW. Un caso de liquen aureus
infantil. Ann Dermatol 2009; 21: 393.
Yazdi
AS, Mayser P, Sander CA. Liquen aureus con células T clonales en un niño
posiblemente inducido por el consumo regular de una bebida energética. J Cutan
Pathol 2008; 35: 960.
Fujita
H, Iguchi M, Ikari Y, Asahina A. Lichen aureus en la espalda de una niña de 6
años. J Dermatol 2007; 34: 148.
Rubio
FA, Robayna G, Herranz P, et al. Liquen aureus abdominal en un niño. Pediatr
Dermatol 1997; 14: 411.
Saito R,
Matsuoka Y. Dermatosis purpúrica granulomatosa pigmentada. J
Dermatol 1996; 23: 551.
Wong WR, Kuo TT, Chen MJ, Chan HL. Variante granulomatosa de la dermatosis
purpúrica pigmentada crónica: informe de dos casos. Br J
Dermatol 2001; 145: 162.
Lin WL, Kuo TT, Shih PY, et al. Variante granulomatosa de las
dermatosis purpúricas pigmentadas crónicas: informe de cuatro casos nuevos y
una asociación con hiperlipidemia. Clin Exp Dermatol 2007; 32: 513.
Kerns
MJ, Mallatt BD, Shamma HN. Púrpura pigmentada granulomatosa: una variante
histológica inusual. Am J Dermatopathol 2009; 31:77.
Lee SH, Kwon JE, Lee KG, Roh MR. Variante granulomatosa de la
dermatosis purpúrica pigmentada crónica asociada con hiperlipidemia. J Eur Acad
Dermatol Venereol 2010; 24: 1243.
Kaplan
J, Burgin S, Sepehr A. Púrpura pigmentada granulomatosa: informe de un caso y
revisión de la literatura. J Cutan Pathol 2011; 38: 984.
Satoh T,
Yokozeki H, Nishioka K. Púrpura pigmentada crónica asociada con infección
odontogénica. J Am Acad Dermatol 2002; 46: 942.
Capo U,
Selle C, Isbruch K, Isbruch K. Púrpura recurrente debido a la enfermedad de
Schamberg relacionada con el alcohol y su asociación con las inmunoglobulinas
séricas: una observación longitudinal de un bebedor pesado. J Med Case Rep
2016; 10: 301.
Pratt M,
Taraska V. Los tintes azules dispersos 106 y 124 son causas comunes de
dermatitis textil y deben servir como alérgenos de detección para esta
afección. Am J Contact Dermat 2000; 11:30.
van der Veen JP, Neering H, de Haan P, Bruynzeel DP. Dermatitis pigmentada de la ropa
purpúrica debida a Disperse Blue 85. Dermatitis de contacto 1988; 19: 222.
Foti C,
Elia G, Filotico R, Angelini G. Dermatitis de la ropa purpúrica debida a
Disperse Yellow 27. Dermatitis de contacto 1998; 39: 273.
Lazarov
A, Córdoba M. La prueba del parche purpúrico en pacientes con dermatitis
alérgica de contacto por colorantes azoicos. Dermatitis de contacto 2000;
42:23.
Shimunes
E. Dermatitis de contacto alérgica purpúrica a parafenilendiamina. Dermatitis
de contacto 1978; 4: 225.
Roed-Petersen
J, Clemmensen OJ, Menné T, Larsen E. Dermatitis de contacto purpúrica por
productos químicos de caucho negro. Dermatitis de contacto 1988; 18: 166.
Schmidt
H, Larsen FS, Larsen PO, Søgaard H. Reacción petequial después de la prueba de
parche con cobalto. Dermatitis de contacto 1980; 6:91.
Laurberg
G, Christiansen JV. Dermatitis de contacto alérgica purpúrica a resina epoxi.
Dermatitis de contacto 1984; 11: 186.
Koch P,
HP Baum, John S. Reacción de prueba del parche purpúrico y venulitis por
metacrilato de metilo en una prótesis dental. Dermatitis de contacto 1996; 34:
213.
de
Waard-van der Spek FB, Oranje AP. La púrpura causada por Emla es de origen
tóxico. Dermatitis de contacto 1997; 36:11.
van
Joost T, van Ulsen J, Vuzevski VD, y col. Dermatitis de contacto purpúrica con
peróxido de benzoilo. J Am Acad Dermatol 1990; 22: 359.
Abeck D, Gross GE, Kuwert C, et al. Púrpura pigmentaria progresiva
inducida por acetaminofén (enfermedad de Schamberg). J Am Acad Dermatol 1992;
27: 123.
Kwon SJ,
Lee CW. Erupciones purpúricas figuradas en el tronco: erupciones inducidas por
acetaminofén. J Dermatol 1998; 25: 756.
Lipsker
D, Cribier B, Heid E, Grosshans E. [Linfoma cutáneo que se manifiesta como
capilares purpúricos pigmentados]. Ann Dermatol Venereol 1999; 126: 321.
Peterson
WC Jr, Manick KP. Erupciones purpúricas asociadas con el uso de carbromal y
meprobamato. Arch Dermatol 1967; 95:40.
Nishioka K, Katayama I, Masuzawa M, et al. Púrpura pigmentada crónica
inducida por fármacos. J Dermatol 1989; 16: 220.
Voelter
WW. Reacción similar a la dermatosis purpúrica pigmentada al fluorouracilo
tópico. Arch Dermatol 1983; 119: 875.
Adams
BB, Gadenne AS. Dermatosis purpúrica pigmentada inducida por glipizida. J Am
Acad Dermatol 1999; 41: 827.
Tsao H,
Lerner LH. Erupción purpúrica pigmentada asociada con inyección de acetato de
medroxiprogesterona. J Am Acad Dermatol 2000; 43: 308.
Koçak AY, Akay BN, Heper AO. Dermatosis purpúrica pigmentada
inducida por sildenafil. Cutan Ocul Toxicol 2013; 32:91.
Nishioka
K, Sarashi C, Katayama I. Púrpura pigmentada crónica inducida por sustancias
químicas. Clin Exp Dermatol 1980; 5: 213.
Kaplan
R, Meehan SA, Leger M. Un caso de telangiectodos de púrpura anular inducidos
por isotretinoína de Majocchi y revisión de dermatosis purpúrica pigmentada
inducida por sustancias. JAMA Dermatol 2014; 150: 182.
Schetz
D, Kocić I. Una nueva reacción adversa a los medicamentos: la enfermedad de
Schamberg causada por la administración de amlodipino: un informe de un caso.
Br J Clin Pharmacol 2015; 80: 1477.
Miraglia
E, Bianchini D, Panetta C, et al. Un caso de dermatitis purpúrica pigmentada
liquenoide de Gougerot-Blum después de amoxicilina. G Ital Dermatol Venereol
2019.
Macquarrie
EK, Pasternak S, Torok M, et al. Dermatitis purpúrica pigmentada persistente:
variante granulomatosa. J Cutan Pathol 2011; 38: 979.
Tato BP,
Marinero Escobedo S, Pérez González YC, et al. Variante granulomatosa de la
dermatosis purpúrica pigmentada. Am J Dermatopathol 2012; 34:
746.
Gupta G, Holmes SC, Spence E, Mills PR. Capillaritis asociada con el
tratamiento con interferón alfa de la infección crónica por hepatitis C. J Am
Acad Dermatol 2000; 43: 937.
Dessoukey
MW, Abdel-Dayem H, Omar MF, Al-Suweidi NE. Dermatosis purpúrica pigmentada y
perfil de hepatitis: un informe sobre 10 pacientes. Int J Dermatol 2005; 44:
486.
Sawamura
D, Ohata Y, Shibaki A, Shimizu H. Dermatosis purpúrica pigmentada en
crioglobulinemia mixta asociada con artritis reumatoide e infección por
hepatitis C. Acta Derm Venereol 2005; 85: 460.
Raslan
HM, Ezzat WM, Abd El Hamid MF, y col. Manifestaciones cutáneas de la infección
crónica por el virus de la hepatitis C en El Cairo, Egipto. East
Mediterr Health J 2009; 15: 692.
Ehsani AH, Ghodsi SZ, Nourmohammad-Pour P, et al. Dermatosis púrpura purificada
pigmentada y hepatitis viral: un estudio de casos y controles. Australas
J Dermatol 2013; 54: 225.
Magro CM, Schaefer JT, Crowson AN, et al. Dermatosis purpúrica pigmentada:
clasificación por perfiles fenotípicos y moleculares. Am J Clin
Pathol 2007; 128: 218.
Shelley WB, Swaminathan R, Shelley ED. Liquen aureus: un tatuaje de
hemosiderina asociado con la incompetencia de las venas perforantes. J Am Acad
Dermatol 1984; 11: 260.
Aiba S,
Tagami H. Estudios inmunohistológicos en la enfermedad de Schamberg. Evidencia
de reacción inmune celular en piel lesionada. Arch Dermatol 1988; 124: 1058.
Ghersetich I, Lotti T, Bacci S, et al. Infiltrado celular en púrpura
progresiva pigmentada (enfermedad de Schamberg): inmunofenotipo, receptores de
adhesión y relaciones intercelulares. Int J Dermatol 1995; 34: 846.
Krizsa
J, Hunyadi J, Dobozy A. Tratamiento con PUVA de dermatitis liquenoide purpúrica
pigmentada (Gougerot-Blum). J Am Acad Dermatol 1992; 27: 778.
Iwatsuki K, Aoshima T, Tagami H, et al. Estudio de inmunofluorescencia en
púrpura pigmentosa crónica. Acta Derm Venereol 1980; 60: 341.
Schamberg
JF. Una peculiar enfermedad pigmentaria progresiva de la piel. Fr. J. Dermatol
1901; 13: 1.
Roehm
HR. Dermatosis pigmentaria progresiva (Schamberg). Clin Pediatr (Phila) 1968;
7: 307.
Hersh
CS, Shwayder TA. Púrpura pigmentaria progresiva unilateral (enfermedad de
Schamberg) en un niño de 15 años. J Am Acad Dermatol 1991; 24: 651.
Draelos
ZK, Hansen RC. La púrpura de Schamberg en niños: estudio de caso y revisión de
la literatura. Clin Pediatr (Phila) 1987; 26: 659.
Kerem E,
Branski D, Gross-Kieselstein E, y col. Púrpura pigmentada crónica: reporte de
un caso de enfermedad de Schamberg. Clin Pediatr (Phila) 1987; 26: 657.
Nichamin
SJ, Brough AJ. Púrpura pigmentaria progresiva crónica. Púrpura anular
telangiectodes de Majocchi-Schamberg. Am J Dis Child 1968; 116: 429.
Majocchi
D. Sopra una dermatosi telangiectodo no ancora descrita: púrpura anular. G Ital
Mal Ven 1896; 31: 263.
Wang A,
Shuja F, Chan A, Wasko C. Telangiectodos de púrpura anular unilateral de
majocchi en un hombre anciano: una presentación atípica. Dermatol Online J
2013; 19: 19263.
Hoesly
FJ, Huerter CJ, Shehan JM. Purpura annularis telangiectodes de Majocchi:
reporte de caso y revisión de la literatura. Int J Dermatol 2009; 48: 1129.
Kim HJ,
Skidmore RA, Woosley JT. Púrpura pigmentada sobre las extremidades inferiores.
Purpura annularis telangiectodes de Majocchi. Arch Dermatol 1998; 134: 1477,
1480.
Hale EK.
Purpura annularis telangiectodes de Majocchi. Dermatol Online J 2003; 9:17.
Gougerot
H, Blum P. Purpura angioscleux prurigeneux avec elements lichenoides. Bull
Soc Franc Derm Syph 1925; 32: 161.
Wong RC, Solomon AR, Field SI, Anderson TF. Dermatitis liquenoide purpúrica
pigmentada de Gougerot-Blum que imita el sarcoma de Kaposi. Cutis 1983; 31:
406.
Marten
RH. Caso para el diagnóstico. Trans St. John's Hosp Dermatol Soc 1958; 30:98.
Calnan
CD. Liquen aureus. Fr. J. Dermatol 1960; 72: 373.
Precio
ML, Jones EW, Calnan CD, MacDonald DM. Liquen aureus: una forma persistente
localizada de dermatitis purpúrica pigmentada. Br J Dermatol 1985; 112: 307.
Mishra
D, Maheshwari V. Liquen aureus segmentario en un niño. Int J
Dermatol 1991; 30: 654.
Leal-Khouri S, Sherman LD, Mallory SB. Erupción segmentaria en una niña
de 8 años. Pediatr Dermatol 1992; 9: 154.
Abramovits
W, Landau JW, Lowe NJ. Un informe de dos pacientes con liquen aureus. Arch
Dermatol 1980; 116: 1183.
Ruiz-Esmenjaud
J, MV Dahl. Liquen aureus segmentario: inicio asociado con trauma y pubertad. Arch
Dermatol 1988; 124: 1572.
Lee HW, Lee DK, Chang SE y col. Liquen aureus segmentario:
terapia combinada con pentoxifilina y prostaciclina. J Eur Acad Dermatol
Venereol 2006; 20: 1378.
Graham
RM, inglés JS, Emmerson RW. Liquen aureus - un estudio de doce casos. Clin Exp
Dermatol 1984; 9: 393.
MOSTO
SJ, CASALA AM. ANGIODERMATITIS PRURIGINOSA DISEMINADA: PURPURA PICANTE. Arch
Dermatol 1965; 91: 351.
DJ
Pravda, Moynihan GD. Picazón púrpura. Cutis 1980; 25: 147.
Riordan
CA, Darley C, Markey AC, et al. Capilarritis lineal unilateral. Clin Exp
Dermatol 1992; 17: 182.
Ma HJ,
Zhao G, Liu W, y col. Capilarritis lineal unilateral: dos casos inusuales
chinos. Eur J Dermatol 2007; 17: 160.
Mar A,
Fergin P, Hogan P. Erupción purpúrica pigmentada unilateral. Australas J
Dermatol 1999; 40: 211.
Taketuchi
Y, Chinen T, Ichikawa Y, Ito M. Dos casos de dermatosis purpúrica pigmentada
unilateral. J Dermatol 2001; 28: 493.
Abdullah
L, Abbas O. Dermacase. ¿Puedes identificar esta condición? Capilarritis lineal
unilateral. Can Fam Physician 2011; 57: 563.
Battle
LR, Shalin SC, Gao L. Dermatosis purpúrica pigmentada granulomatosa. Clin Exp
Dermatol 2015; 40: 387.
Hanson
C, Fischer R, Fraga G, Rajpara A. Dermatosis purpúrica granulomatosa
pigmentada: una variante inusual asociada con hiperlipidemia. Dermatol Online J
2014; 21)
MacKenzie
AI, Biswas A. Dermatosis purpúrica granulomatosa pigmentada: informe de un caso
con presentación clínica atípica que incluye hallazgos dermatoscópicos. Am J
Dermatopathol 2015; 37: 311.
Carvajal
D, Quiroz C, Morales C, Fernández J. Dermatosis purpúrica pigmentada
granulomatosa: informe de un caso latinoamericano con distribución de
blascoides. An Bras Dermatol 2019; 94: 582.
Weedon
D. El patrón de reacción vasculopática. En:
Weedon's Skin Pathology, 3ª ed., Elsevier Limited, 2010. p.195.
Fishman
HC. Dermatitis liquenoide purpúrica pigmentada de Gougerot-Blum. Cutis 1982;
29: 260.
Ozkaya
DB, Emiroglu N, Su O, et al. Hallazgos dermatoscópicos de dermatosis purpúrica
pigmentada. An Bras Dermatol 2016; 91: 584.
Toro JR,
Sander CA, LeBoit PE. La dermatitis purpúrica pigmentada persistente y la
micosis fungoide: ¿simulante, precursor o ambos? Un estudio por microscopía
óptica y métodos moleculares. Am J Dermatopathol 1997; 19: 108.
Lor P,
Krueger U, Kempf W, et al. Reordenamiento monoclonal de la cadena gamma del
receptor de células T en dermatitis purpúrica pigmentada liquenoide de
gougerot-blum que responde a la terapia tópica con corticosteroides
Dermatología 2002; 205: 191.
Rudolph RI. Liquen aureus. J Am Acad Dermatol 1983; 8:
722.
Risikesan J, Sommerlund M, Ramsing M, et al. Tratamiento tópico exitoso de la
dermatitis liquenoide purpúrica pigmentada de Gougerot-Blum en un paciente
joven: informe de un caso y resumen de las dermatosis purpúricas pigmentadas
más comunes. Representante de Caso Dermatol 2017; 9: 169.
Fathy H,
Abdelgaber S. Tratamiento de dermatosis purpúricas pigmentadas con UVB de banda
estrecha: un informe de seis casos. J Eur Acad Dermatol Venereol 2011; 25: 603.
Lasocki
AL, Kelly RI. Terapia UVB de banda estrecha como tratamiento efectivo para la enfermedad
de Schamberg. Australas J Dermatol 2008; 49:16.
Gudi VS,
White MI. Púrpura pigmentada progresiva (enfermedad de Schamberg) que responde
a la terapia con ultravioleta B TL01. Clin Exp Dermatol 2004; 29: 683.
Karadag
AS, Bilgili SG, Onder S, Calka O. Dos casos de púrpura eccematídica de Doucas y
Kapetanakis que responden al tratamiento ultravioleta B de banda estrecha.
Photodermatol Photoimmunol Photomed 2013; 29:97.
Dhali
TK, Chahar M, Haroon MA. La fototerapia como tratamiento efectivo para la enfermedad
de Majocchi - reporte de caso. An Bras Dermatol 2015; 90:96.
Can B,
Turkoglu Z, Kavala M, et al. Tratamiento exitoso de la enfermedad de Schamberg
infantil generalizada con terapia ultravioleta B de banda estrecha. Photodermatol
Photoimmunol Photomed 2011; 27: 216.
Kocaturk E, Kavala M, Zindanci I, et al. Tratamiento de banda estrecha UVB
de dermatitis liquenoide purpúrica pigmentada (Gougerot-Blum). Photodermatol
Photoimmunol Photomed 2009; 25:55.
Lotti T,
Ghersetich I, Panconesi E. ¿Por qué debemos usar el tratamiento con PUVA en la
dermatitis liquenoide purpúrica pigmentada? J Am Acad Dermatol 1994; 30: 145.
Seckin
D, Yazici Z, Senol A, Demircay Z. Un caso de enfermedad de Schamberg que
responde dramáticamente al tratamiento con PUVA. Photodermatol
Photoimmunol Photomed 2008; 24:95.
Ling TC, Goulden V, Goodfield MJ. Terapia PUVA en
liquen aureus. J Am
Acad Dermatol 2001; 45: 145.
Panda S,
Malakar S, Lahiri K.Oral pentoxifilina versus betametasona tópica en la
enfermedad de Schamberg: un ensayo aleatorio comparativo de grupo paralelo
cegado por investigadores. Arch Dermatol 2004; 140: 491.
Kano Y,
Hirayama K, Orihara M, Shiohara T. Tratamiento exitoso de la enfermedad de
Schamberg con pentoxifilina. J Am Acad Dermatol 1997; 36: 827.
Wahba-Yahav
AV. Púrpura de Schamberg: asociación con antigenemia de superficie persistente
de hepatitis B y tratamiento con pentoxifilina. Cutis 1994; 54: 205.
Basak
PY, Ergin S. ¿Debería considerarse la pentoxifilina como un tratamiento eficaz
para la enfermedad de Schamberg? J Am Acad Dermatol 2001; 44: 548.
Burkhart
CG, Burkhart KM. Pentoxifilina para la enfermedad de Schamberg. J Am Acad
Dermatol 1998; 39: 298.
Tamaki
K, Yasaka N, Osada A, et al. Tratamiento exitoso de la dermatosis purpúrica
pigmentada con griseofulvina. Br J Dermatol 1995; 132: 159.
Agrawal
SK, Gandhi V, Bhattacharya SN. Dobesilato de calcio (Cd) en dermatosis
purpúrica pigmentada (PPD): una evaluación piloto. J Dermatol 2004; 31:98.
Geller
M. Beneficio de la colchicina en el tratamiento de la enfermedad de Schamberg.
Ann Alergia Asma Immunol 2000; 85: 246.
Cavalcante
MLLL, Masuda PY, Brito FF, et al. Enfermedad de Schamberg: reporte de caso con
éxito terapéutico mediante el uso de colchicina. An Bras Dermatol 2017; 92:
246.
Murota
H, Katayama I. Liquen aureus que responde al tratamiento tópico con tacrolimus.
J Dermatol 2011; 38: 823.
Kim SK,
Kim EH, Kim YC. Tratamiento de la dermatosis purpúrica pigmentada con terapia
fotodinámica tópica. Dermatología 2009; 219: 184.
Hong DK,
Chang IK, Lee Y, et al. Tratamiento del liquen aureus segmentario con un láser
de colorante pulsado: nuevas opciones de tratamiento para el liquen aureus. Eur
J Dermatol 2013; 23: 891.
Arreola
Jauregui IE, López Zaldo JB, Huerta Rivera G, et al. Un caso de liquen aureus
tratado con éxito con láser de colorante pulsado de longitud de onda de 595 nm.
J Cosmet Dermatol 2020; 19: 657.
Okada K,
Ishikawa O, Miyachi Y. Púrpura pigmentosa crónica tratada con éxito con
ciclosporina oral A. Br J Dermatol 1996; 134: 180.
Reinhold
U, Seiter S, Ugurel S, Tilgen W. Tratamiento de púrpura pigmentada progresiva
con bioflavonoides orales y ácido ascórbico: un estudio piloto abierto en 3
pacientes. J Am Acad Dermatol 1999; 41: 207.
Schober SM, Peitsch WK, Bonsmann G, y col. El tratamiento temprano con
rutósido y ácido ascórbico es altamente efectivo para la dermatosis purpúrica
pigmentada progresiva. J Dtsch Dermatol Ges 2014; 12: 1112.
Barnhill
RL, Braverman IM. Progresión de erupciones pigmentadas de tipo púrpura a
micosis fungoide: informe de tres casos. J Am Acad Dermatol 1988; 19:25.
Martínez
W, del Pozo J, Vázquez J, et al. Linfoma cutáneo de células T que se presenta
como erupción diseminada, pigmentada, similar a la púrpura. Int J Dermatol
2001; 40: 140.
Ugajin
T, Satoh T, Yokozeki H, Nishioka K. Micosis fungoides que se presentan como
erupción purpúrica pigmentada. Eur J Dermatol 2005; 15: 489.
Kazakov
DV, Burg G, Kempf W. Espectro clinicopatológico de micosis fungoide. J
Eur Acad Dermatol Venereol 2004; 18: 397.
Georgala S, Katoulis AC, Symeonidou S, et al. Erupción purpúrica pigmentada
persistente asociada con micosis fungoide: reporte de un caso y revisión de la
literatura. J Eur Acad Dermatol Venereol 2001; 15:62.
Puddu P,
Ferranti G, Frezzolini A y col. Erupción pigmentada similar a la púrpura como
signo cutáneo de micosis fungoide con púrpura autoinmune. J
Am Acad Dermatol 1999; 40: 298.
Cather JC, Farmer A, Jackow C, et al. Presentación inusual de micosis
fungoide como púrpura pigmentada con timoma maligno. J Am Acad Dermatol 1998;
39: 858.
Crowson
AN, Magro CM, Zahorchak R. Púrpura pigmentaria atípica: un estudio clínico,
histopatológico y genotípico. Hum Pathol 1999; 30: 1004.
Ladrigan
MK, Poligone B. El espectro de dermatosis purpúrica pigmentada y micosis
fungoide: discrasia atípica de células T. Cutis 2014; 94: 297.















