Masculino de 85 años diabético, hipertenso pescador por
muchos años acude a cónsulta hoy 26 de Diciembre , con lesiones cutáneas de Varios
años asintomáticas.
Se solicitó biopsia cuyo resultado fue poroqueratosis.
Presentó
Dr. Roberto Miranda Chapa
Dermatólogo. Docente de Postgrado de Medicina Familiar y Medicina Interna.
Hospital General Regional 6 del IMSS Tampico
Tamaulipas Mexico.
POROQUERATOSIS
INTRODUCCIÓN
La poroqueratosis es un trastorno de queratinización raro,
adquirido o heredado que se caracteriza por una o más máculas o parches
atróficos, cada uno rodeado por un distintivo borde hiperqueratótico en forma
de cresta llamado "lámina cornoidea" ( imagen 6 ) [ 1,2 ]. Existen
múltiples variantes clínicas de poroqueratosis. La transformación maligna
ocurre en una minoría de casos.
Imagen 6: Poroqueratosis
Máculas eritematosas bien definidas con un borde periférico
de escama ("lámina cornoidea").
Aunque la vigilancia clínica para la transformación maligna
es suficiente para el tratamiento de la mayoría de los pacientes con poroqueratosis,
los pacientes que están preocupados por la aparición de lesiones o que tienen
síntomas asociados, como prurito o dolor, pueden desear una intervención
terapéutica.
EPIDEMIOLOGÍA
La poroqueratosis es una enfermedad rara. Se desconocen la
incidencia y prevalencia exactas. Suele ocurrir en adultos o adultos mayores
con ligero predominio masculino, pero se han informado casos pediátricos [ 3 -
7 ].
CLASIFICACIÓN
Las variantes de poroqueratosis más comúnmente descritas
incluyen:
- Poroqueratosis actínica superficial diseminada (DSAP1, MIM #
175900, 616063, 614714, 616631)
- Poroqueratosis superficial diseminada (DSP, MIM # 175900,
616631)
- Poroqueratosis de Mibelli (MIM # 175800)
- Poroqueratosis palmaris y plantar diseminada (PPPD, MIM #
175850)
- Poroqueratosis punteada (a veces considerada una variante de
PPPD)
- Poroqueratosis psicotropica
- Pororatosis genitoglutea.
La mayoría de los tipos clínicos de poroqueratosis pueden
manifestarse en formas localizadas o generalizadas; sin embargo, la
superposición entre las formas y las variantes clínicas no son infrecuentes.
PATOGENESIS
Se cree que la proliferación clonal de los queratinocitos
epidérmicos anormales explica las manifestaciones clínicas de la poroqueratosis.
Sin embargo, el camino que conduce a este proceso sigue siendo desconocido. Se
han propuesto una variedad de factores como posibles contribuyentes, incluida
la susceptibilidad genética, la radiación ultravioleta y el estado inmunitario.
Genética: los
defectos genéticos hereditarios o esporádicos probablemente juegan un papel
importante en la poroqueratosis. Los estudios en casos familiares de poroqueratosis
actínica superficial diseminada (DSAP) respaldan un patrón de herencia
autosómico dominante con penetrancia incompleta [ 8 ].
Se han identificado cuatro posibles loci cromosómicos para
DSAP, incluidos DSAP1 (12q23.2-24.1), DSAP2 (15q25.1-26.1), DSAP3
(1p31.3-p31.1) y DSAP4 (16q24.1-24.3) [ 9-12 ]. En estudios chinos , se han
detectado mutaciones en los genes de la ruta de la fosfomevalonato quinasa, a
saber, mevalonato descarboxilasa ( MVD ), mevalonato quinasa ( MVK ),
fosfomevalonato quinasa ( PMVK ) y farnesil difosfato sintasa ( FDPS ), en
algunos pacientes con poroqueratosis [ 13, 14 ] Hay más de 200 mutaciones
identificadas hasta ahora [ 14-17] Se informó que al menos una mutación en los
genes de la ruta del mevalonato se encontró en hasta el 98 por ciento de los
casos familiares de poroqueratosis esporádica y en más del 70 por ciento [ 18
].
Los genes de la vía del mevalonato están involucrados en la
biosíntesis de los isoprenoides, que son precursores de múltiples sustancias
biológicamente importantes, como el colesterol, y están indirectamente
involucrados en eventos biológicos, como el crecimiento celular de la
diferenciación. Por lo tanto, se presume que las mutaciones en la ruta del
mevalonato dan como resultado la acumulación de metabolitos celulares
anormales, la retención de núcleos celulares en la epidermis y la formación de
la lámina cornoidea [ 18,19 ]. Además, la inhibición de las enzimas dentro de
la ruta del mevalonato conduce a una disminución de la expresión del marcador
de diferenciación de queratinocitos involucrina, p53 y Notch1, lo que respalda
aún más la relación causal entre las mutaciones en la ruta del mevalonato y el
desarrollo de poroqueratosis [ 20 ].
Los casos familiares de poroqueratosis superficial
diseminada (DSP) y poroqueratosis plantar palmaris y diseminada (PPPD)
demuestran patrones de herencia autosómica dominante. Los loci vinculados a DSP
y PPPD están en el cromosoma 18p11.3 y el cromosoma 12q24.1-24.2, respectivamente
[ 21-23 ].
Las formas focales de poroqueratosis, como la poroqueratosis
de Mibelli y la poroqueratosis lineal, pueden ocurrir como consecuencia del mosaicismo.
La aparición de poroqueratosis lineal o poroqueratosis de Mibelli en pacientes
con DSAP se ha atribuido a esta teoría [ 8,24 ]. En tales casos, la pérdida
focal de heterocigosidad a través de mutaciones somáticas puede dar lugar a
manifestaciones clínicas prominentes y localizadas que caracterizan a la poroqueratosis
lineal y a la poroqueraratosis de Mibelli, mientras que las manifestaciones
relativamente más leves de DSAP aparecen en una distribución generalizada [
8,24 ].
Radiación ultravioleta: la
exposición a la radiación ultravioleta puede contribuir al desarrollo de
poroqueratosis. Esta afirmación está respaldada por la observación de que DSAP
ocurre preferentemente en áreas de piel expuesta y en individuos con
antecedentes de exposición prolongada al sol. Además, la administración de luz
ultravioleta artificial en entornos experimentales y terapéuticos ha resultado
en la inducción de lesiones DSAP [ 25-28 ].
A pesar de estos hallazgos, el papel definitivo de la radiación
ultravioleta en la poroqueratosis sigue siendo incierto. La mejora en DSAP
después del tratamiento con psoraleno más la terapia con ultravioleta A (PUVA)
se ha documentado en un paciente [ 29 ]. El relativo respeto de la cara en DSAP
también plantea preguntas sobre la designación de la radiación ultravioleta como
factor contribuyente.
Inmunosupresión: la
inmunosupresión o inmunodeficiencia inducida por fármacos en el contexto de
enfermedades no malignas o neoplasias hematológicas puede aumentar el riesgo de
poroqueratosis [ 30-36 ]. Las estimaciones de la prevalencia de poroqueratosis
en receptores de trasplantes de órganos inmunosuprimidos varían desde menos del
1 por ciento hasta el 11 por ciento [ 32,35 ].
La poroqueratosis ha ocurrido 3 semanas a 14 años después
del inicio de la inmunosupresión sistémica [ 35,37 ]. DSP y poroqueratosis de
Mibelli son las variantes más comúnmente asociadas [ 38 ]. Se ha descrito una
forma de porokeratosis de rápida evolución, llamada porokeratosis diseminada
eruptiva, en pacientes mayores, en asociación con cánceres sólidos o terapia
inmunosupresora [ 39 ].
El DSP de nueva aparición y la poroqueratosis de Mibelli se
han informado en pacientes con infección por VIH conocida y pueden indicar un
empeoramiento de la inmunodeficiencia [ 31,40 ].
Un papel contribuyente para la inmunosupresión está
respaldado por los informes de remisión de poroqueratosis después del cese de
la terapia inmunosupresora en dos pacientes [ 41,42 ]. El desarrollo de poroqueratosis
en sitios de uso potente a largo plazo de corticosteroides tópicos en otro
paciente también sugiere un papel para la inmunosupresión [ 43 ].
Otros: los factores
adicionales que se han asociado con la poroqueratosis incluyen radioterapia [
44,45 ], trauma [ 46 ], enfermedad hepática [ 47,48 ], cáncer de órganos
sólidos [ 49,50 ] y enfermedad de Crohn [ 51 ].
PRESENTACIÓN CLÍNICA
La característica definitoria de la poroqueratosis es la
presencia clínica e histopatológica de una lámina cornoidea, que generalmente
se manifiesta como un borde delgado y queratótico en la periferia de una lesión
cutánea ligeramente atrófica ( imagen 6 ). Las lesiones suelen comenzar como
pequeñas pápulas queratóticas que se extienden lenta y centrífugamente [ 38 ].
Se han descrito múltiples variantes clínicas. La poroqueratosis
actínica superficial diseminada (DSAP) se acepta generalmente como la variante
más común, seguida de la poroqueratosis de Mibelli. En una serie de 94
pacientes en Singapur, la poroqueratosis clásica de Mibelli, DSAP, la poroqueratosis
superficial diseminada (DSP) y la poroqueratosis lineal representaron el 56,
18, 11 y 13 por ciento de los casos de poroqueratosis, respectivamente [ 5 ].
POROQUERATOSIS ACTÍNICA DISEMINADA SUPERFICIAL (DSAP)
Es el tipo más común de
poroqueratosis. El DSAP ocurre con mayor frecuencia en mujeres que en hombres,
con una proporción estimada de mujeres a hombres de aproximadamente 1.8: 1 [ 38
]. El inicio de la enfermedad generalmente ocurre en la tercera o cuarta década
de la vida, y los pacientes frecuentemente reportan un historial de exposición
extensa a la radiación ultravioleta natural o artificial. Puede empeorar la
enfermedad durante los meses de verano [ 38 ].
DSAP se caracteriza por máculas eritematosas, del color de
la piel o hiperpigmentadas, bien definidas, que típicamente tienen menos de 1
cm de diámetro ( imágenes 7,8 y 9 ). La lámina cornoidea es a menudo sutil y se
manifiesta como una cresta fina, periférica y queratótica. La distribución de
las lesiones generalmente involucra las superficies extensoras de los brazos,
piernas, hombros o espalda, con preservación de las palmas y las plantas de los
pies.
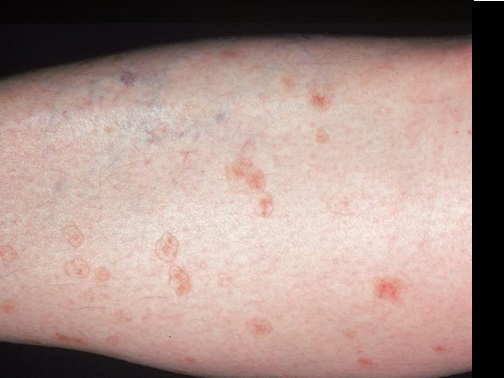
Imagen 7: Porqueratosis actínica superficial diseminada
Múltiples máculas eritematosas con bordes periféricos
queratósicos están presentes en la parte inferior de las piernas.
Imagen 8: Poroqueratosis actínica superficial diseminada.
Múltiples máculas eritematosas bien definidas con un borde
periférico de escala están presentes en la pierna de este paciente con poroqueratosis
actínica superficial diseminada.
Imagen 9: Poroqueratosis actínica superficial diseminada
Múltiples máculas eritematosas con bordes periféricos
queratóticos están presentes en la extremidad.
Las lesiones faciales, un hallazgo menos común, ocurren en
aproximadamente el 15 por ciento de los pacientes [ 52 ]. Una variante menos
común (DSAP inflamatorio) se presenta como lesiones eritematosas, acompañadas
de inflamación y prurito severo [ 53 ].
El número de lesiones cutáneas en DSAP es variable. Sólo
unos pocos o varios cientos de lesiones pueden estar presentes [ 29 ]. El
aspecto cosmético de DSAP es una preocupación común para los pacientes con este
trastorno y puede ser particularmente molesto para las personas que
frecuentemente usan faldas o pantalones cortos que exponen la parte inferior de
las piernas. Las lesiones cutáneas a menudo son asintomáticas, pero se estima
que se producen prurito o sensaciones punzantes en un tercio a la mitad de los
pacientes [ 29,38 ].
poroqueratosis diseminada superficial
La poroqueratosis diseminada superficial (DSP) se asemeja a
DSAP pero carece fotodistribution, apareciendo tanto en sitios que no están
expuestos al sol como en los expuesta al sol. Al igual que DSAP, las palmas y
las plantas generalmente están respetadas.
A diferencia del DSAP, el DSP a menudo se desarrolla en la
infancia, más comúnmente entre las edades de 5 y 10 años [ 38 ]. Como se señaló
anteriormente, el DSP también puede ocurrir en asociación con la
inmunosupresión [ 54-56 ]
El término "POROQUERATOSIS DISEMINADA ERUPTIVA" se
ha propuesto para la poroqueratosis diseminada aguda de inicio en adultos, que
tiene una preponderancia masculina y puede mostrar una morfología variable [ 39
]. La mayoría de los pacientes con DSP de inicio en adultos están
inmunodeprimidos o tienen una neoplasia maligna interna coexistente.
POROQUERATOSIS DE MIBELLI
La poroqueratosis de
Mibelli es el segundo tipo más común de poroqueratosis [ 3 ]. El trastorno a
menudo comienza en la infancia y afecta a los hombres aproximadamente el doble
de frecuencia que las mujeres [ 38 ]. Ocasionalmente, las lesiones se
desarrollan durante la edad adulta, a menudo en asociación con inmunosupresión
[ 31,33,35,57 ].
La poroqueratosis de Mibelli generalmente comienza como una
pápula pequeña, asintomática o ligeramente pruriginosa que se expande lentamente
a lo largo de los años. Un crecimiento más rápido puede ocurrir en el contexto
de inmunosupresión. Las lesiones bien desarrolladas generalmente tienen unos
pocos centímetros de diámetro y, en raras ocasiones, las lesiones crecen de 10
a 20 cm de tamaño. El término "poroqueratosis gigante" se ha
utilizado para describir estas lesiones excepcionalmente grandes [ 58,59 ].
La ubicación clásica para la poroqueratosis de Mibelli es en
una extremidad. Sin embargo, las lesiones pueden ocurrir en cualquier sitio,
incluidas las palmas, las plantas de los pies, los genitales o las membranas
mucosas [ 38,60,61 ]. Los pacientes generalmente presentan una lesión solitaria
o algunas lesiones distribuidas asimétricamente. Ocasionalmente, se producen
lesiones generalizadas [ 24,58,62 ]. La participación de las porciones distales
de los dedos de manos y pies puede provocar distrofia y / o pérdida completa de
la uña [ 63 ].
La lámina cornoidea a menudo es prominente en lesiones de
poroqueratosis de Mibelli. El borde de la lesión suele ser mayor de 1 mm de
altura, y generalmente se ve un surco delgado en el centro de la cresta, lo que
causa una apariencia que se asemeja a la Gran Muralla de China ( imagen 10,11,
y 12) ). El centro de la lesión puede estar ligeramente hipopigmentado o
hiperpigmentado, mínimamente escamoso, ligeramente atrófico y sin pelo. Con
poca frecuencia , las lesiones se manifiestan como placas gruesas confluentes,
hiperqueratóticas o verrugosas [ 62,64 ].
Imagen 10: Poroqueratosis de Mibelli
Está presente una placa grande con un centro ligeramente
atrófico y una cresta queratósica prominente, periférica.
Imagen 11: Poroqueratosis de Mibelli.
Múltiples poroqueratosis de lesiones de Mibelli están
presentes
Imagen 12:Poroqueratosis de Mibelli
Está presente un parche eritematoso bien definido con un
borde fino queratótico (lámina cornoidea).
POROQUERATOSIS LINEAL
La poroqueratosis lineal es una forma rara de poroqueratosis
que pueden representar una forma segmentaria de DSAP o DSP [ 8,24 ]. Las
lesiones cutáneas suelen presentarse durante la infancia o la primera infancia,
pero en ocasiones se desarrollan en adultos. Las mujeres son ligeramente más
propensas a verse afectadas que los hombres [ 35 ].
En función de la distribución de las lesiones, la poroqueratosis
lineal se puede subdividir en variantes localizadas, zosteriformes,
sistematizadas o generalizadas ( Imágenes 13 y 14). La variante localizada de
poroqueratosis lineal se presenta como placas únicas o múltiples con bordes
periféricos hiperqueratóticos en una extremidad. Al igual que en la poroqueratosis
de Mibelli, el borde hiperqueratósico a menudo es prominente y se observa un
surco justo dentro de la cresta hiperqueratótica. En la variante zosteriforme,
la configuración de las lesiones generalmente sigue las líneas de Blaschko. La
poroqueratosis lineal sistematizada se presenta como lesiones distribuidas en
las extremidades superiores e inferiores de un lado del cuerpo. En la forma
generalizada de poroqueratosis lineal, las lesiones se encuentran en más de una
extremidad o en el tronco.
Imagen 13: Poroqueratosis lineal
Placas eritematosas lineales con bordes prominentes y
queratósicos están presentes en este paciente con poroqueratosis lineal.
Imagen 14: Poroqueratosis lineal
Placas eritematosas ligeramente atróficas con bordes
periféricos queratósicos.
La poroqueratosis lineal se asocia con un mayor riesgo de
carcinoma de células escamosas [ 65-68 ].
POROQUERATOSIS PLANTAR PALMARIS Y DISEMINADA
La poroqueratosis
plantar palmaris y diseminada (PPPD) es una variante rara de porokeratosis,
heredada en un patrón autosómico dominante, que puede desarrollarse a cualquier
edad [ 3,69 ]. Las lesiones suelen aparecer por primera vez en la adolescencia
o en la edad adulta temprana.
Las manifestaciones iniciales de PPPD son múltiples máculas
pequeñas, relativamente uniformes en las palmas y las plantas que exhiben un
centro ligeramente hiperpigmentado y atrófico y una cresta periférica
mínimamente elevada [ 69-72 ]. Una configuración serpiginosa puede evolucionar
con el tiempo.
Los pacientes con PPPD también pueden desarrollar lesiones
en el tronco y las extremidades que se parecen a las lesiones de DSAP y DSP. La
afectación de la mucosa oral también puede ocurrir como múltiples anillos
opalescentes pequeños, deprimidos y con bordes hiperémicos [ 73 ].
Las lesiones de PPPD pueden ser asintomáticas, pruriginosas
o sensibles a la palpación. Las poroqueratosis en los pies pueden causar
molestias al caminar.
POROQUERATOSIS PUNTEADA
La
poroqueratosis punteada (poroqueratosis punctata palmaris et plantaris)
se caracteriza por múltiples lesiones queratósicas pequeñas, similares a
semillas, en las palmas y las plantas que tienen hallazgos histopatológicos
compatibles con una lámina cornoidea [ 74,75 ]. Algunos autores consideran que
la pororatosis punteada es una especie de fruste de PPPD en lugar de una
entidad separada. (Imagen 14 y 15)
Imagen 14: Poroqueratosis punteada
Imagen 15:
Poroqueratosis punteada.
POROQUERATOSIS PSICOTRÓPICA
La poroqueratosis
psicotrópica es una variante psoriasiforme inusual de la enfermedad, también
descrita como poroqueratosis verrugosa e hipertrófica [ 76-78 ]. Este tipo de
poroqueratosis generalmente se presenta en pacientes masculinos con placas
inflamatorias, queratósicas o verrugosas en las nalgas o la piel genital. La
patología muestra numerosos focos histopatológicos de laminillas cornoides [
76,77,79 ]. Las lesiones de poroqueratosis psicotrópicas a menudo se confunden con psoriasis, liquen
simple crónico o eccema crónico no solo debido a similitudes clínicas ( imagen
16 y 17) sino también debido a lapicazón.
Imagen 16: Poroqueratosis psicotrópica
Placas escamosas están presentes en el pliegue glúteo.
Imagen 17: Poroqueratosis psicotrópica
Placas escamosas están presentes en el pliegue glúteo. Las
laminillas cornoides son visibles en algunas áreas.
POROQUERATOSIS GENITOGLUTEA
El área genital se ve poco frecuentemente afectada por poroqueratosis,
con aproximadamente 50 casos reportados en la literatura [ 80 ]. Las lesiones
poroqueratósicas en el área genital pueden representar una entidad separada o
una forma localizada de otros tipos de poroqueratosis [ 81 ]. La afectación
genital se informa más comúnmente en hombres; sin embargo, también se han publicado
informes de casos de poroqueratosis vulvar.
Se han publicado más de 10 casos de poroqueratosis pene-escrotal
[ 82,83 ]. Se ha informado que la variante poco común de la enfermedad solo
ocurre en hombres jóvenes en su tercera década de vida, con manifestación
clínica en el eje del pene y el escroto anterior acompañada de ardor y picazón
intensos [ 81 ].
OTROS
Otros subtipos de poroqueratosis que se han descrito en la
literatura incluyen la folicular [ 84-87
]; papular pruriginosa eruptiva en el contexto de DSP [ 88-90 ]; gigante; filiforme [ 91 ]; y asociada con craneosinostosis, cierre tardío
de la fontanela, defectos craneales, hipoplasia clavicular, malformaciones
anales y genitourinarias, y síndrome de erupción cutánea (CDAGS) [ 92,93 ].
TRANSFORMACIÓN MALIGNA
La transformación maligna puede ocurrir en pacientes con
todas las variantes principales de poroqueratosis, con la excepción de la
poroqueratosis punteada [ 94 ]. Se estima que ocurre en el 7,5 al 11 por ciento
de los pacientes, con un período promedio de aparición de cáncer de 36 años [
38 ]. El carcinoma de células escamosas es la neoplasia maligna asociada más
frecuente [ 95 ]. El carcinoma de células basales y la enfermedad de Bowen
(carcinoma de células escamosas in situ) también pueden ocurrir.
La poroqueratosis lineal y la poroqueratosis gigante (una
variante de la poroqueratosis de Mibelli) son las variantes más susceptibles a
la transformación maligna, mientras que esta ocurrencia en la porokeratosis
actínica superficial diseminada (DSAP) es menos común [ 94,96,97 ]. Una revisión
de 281 pacientes con poroqueratosis encontró riesgos de transformación maligna
en pacientes con poroqueratosis lineal, porokeratosis plantar palmaris y
diseminada (PPPD), porokeratosis de Mibelli de cualquier tamaño y DSAP de 19,
10, 8 y 3 por ciento, respectivamente [ 94] Sin embargo, como el desarrollo de
DSAP podría depender de la luz ultravioleta y la irradiación ultravioleta
provoca el desarrollo de tumores malignos cutáneos, la presentación concurrente
de DSAP, queratosis actínicas y otros tumores malignos cutáneos no es
infrecuente. El gran tamaño de la lesión, la ubicación en las extremidades y la
larga duración de la lesión son factores adicionales que se han identificado
como factores de riesgo para la transformación maligna [ 38,94,98,99 ].
DIAGNÓSTICO
CLÍNICO
El diagnóstico de poroqueratosis a menudo se puede realizar
basándose únicamente en el examen clínico. La apariencia clínica de una mácula
o parche atrófico con una cresta hiperqueratótica elevada y bien definida sugiere
este trastorno ( imagen 17.1 ). Se puede usar tinta marcadora o yodo para
resaltar la apariencia de la lámina cornoidea en casos sutiles. Las lociones
autobronceadoras que contienen dihidroxiacetona también resaltan la lámina
cornoidea.
Imagen 17.1: Poroqueratosis actínica superficial diseminada
Porqueratosis actínica superficial diseminada
Máculas eritematosas bien definidas con un borde periférico
de escala ("lámina cornoidea").
Las biopsias de piel generalmente no son necesarias. Las
biopsias generalmente se realizan cuando la apariencia de la lesión no es
clásica o cuando existe preocupación por la transformación maligna.
HISTOPATOLOGÍA
Una biopsia por afeitado lo suficientemente profunda como
para incorporar la dermis media es adecuada para el diagnóstico, siempre que
incluya el borde de la lesión (sitio de la lámina cornoidea). Las lesiones
pequeñas pueden eliminarse por completo mediante una escisión por afeitado.
También se puede realizar una biopsia por punción del borde de la lesión.
La característica histopatológica clásica de la poroqueratosis
es la lámina cornoidea, una columna delgada de células paraqueratósicas
apretadas dentro de una invaginación epidérmica llena de queratina ( imagen
18,19 y 20). El vértice de la columna forma un ángulo alejado del centro de la
lesión, y la base de la columna demuestra la interrupción de la capa granular
epidérmica y los queratinocitos disqueratósicos [ 35 ]. Un infiltrado
inflamatorio linfocítico moderado suele estar presente en la dermis papilar. En
ocasiones se observan depósitos amiloides dérmicos [ 76,100,101 ]. La
hiperplasia melanocítica, con o sin hiperpigmentación clínica, se observó en
aproximadamente el 25 por ciento de las muestras en un estudio [ 102 ].
Imagen 18: Histopatología de la poroqueratosis
Se ve una columna delgada y vertical de células
paraqueratóticas dentro de una invaginación epidérmica llena de queratina,
conocida como lámina cornoidea.
Imagen 19: Poroqueratosis
Una columna de poroqueratosis consistente con una lámina
cornoidea está presente en el centro de esta muestra.
Imagen 20:
Una columna de células poroqueratósicas consistentes con una
lámina cornoidea está presente dentro de la dermis.
Aunque la gran mayoría de las muestras de biopsia que
demuestran una lámina cornoidea se derivan de verdaderas poroquratosis, el
hallazgo no es exclusivo de estas lesiones. En raras ocasiones, se pueden
observar láminas cornoides en otros trastornos, como queratosis actínicas,
queratosis seborreicas, verrugas virales y carcinoma basocelular o de células
escamosas [ 103 ].
DERMATOSCOPIA
La dermatoscopia es
una prueba no invasiva que puede ser útil para el diagnóstico si el médico
tiene acceso al equipo y está capacitado para su uso.
En el examen dermatoscópico, la lámina cornoidea aparece
como un borde delgado, blanco y de doble margen que puede tener pigmentación
marrón [ 104-108 ]. El centro atrófico de una lesión a menudo muestra un área
blanca con puntos rojos, glóbulos y líneas que representan vasos capilares [
104 ].
EVALUACIÓN ADICIONAL
Se debe considerar la
posibilidad de inmunosupresión asociada o neoplasia maligna interna
(especialmente neoplasia maligna hematológica) en adultos que presentan
poroqueratosis de Mibelli o poroqueratosis plantaris palmaris y diseminada
(PPPD) de nueva aparición, o exacerbaciones repentinas de un trastorno
poroqueratósico de larga evolución.
DIAGNÓSTICO DIFERENCIAL
La detección de una lámina cornoidea en el examen clínico en
una lesión sospechosa de poroqueratosis suele ser suficiente para el
diagnóstico. Sin embargo, cuando los pacientes presentan hallazgos menos
clásicos, se deben considerar otros trastornos que comparten características
clínicas con poroqueratosis. En tales casos, una biopsia de la lesión para la
evaluación histopatológica es útil para distinguir entre poroqueratosis y otros
trastornos.
Las lesiones de poroqueratosis actínica superficial
diseminada (DSAP) o poroqueratosis superficial diseminada (DSP) clínicamente
pueden parecerse a:
Queratosis actínicas múltiples ( Imagen 21 )
Imagen 21: Queratosis actínica
Las queratosis actínicas múltiples se presentan como máculas
rojas ásperas y escamosas en la piel dañada por el sol.
Queratosis seborreicas maculares ( imagen 22 )
Imagen 22: Queratosis seborreica
Queratosis seborreica superficial que se presenta como una
placa bien demarcada, ligeramente elevada con una apariencia opaca.
Trastornos inflamatorios de la piel, como psoriasis
guttata, pitiriasis rosada, dermatitis numular, tiña del cuerpo y liquen plano.
Las lesiones de poroqueratosis de Mibelli clínicamente
pueden parecerse a:
Carcinoma cutáneo de células escamosas in situ.
Tiña del cuerpo
Granuloma anular
Liquen simple crónico
Hipoqueratosis palmar o plantar circunscrita [ 109 ]
Sarcoidosis [ 110 ]
Las lesiones de poroqueratosis lineal clínicamente pueden
parecerse a:
Verrugas lineales (ver "Verrugas cutáneas (verrugas
comunes, plantares y planas)" )
Liquen estriado.
Nevo epidérmico verrugoso lineal.
Enfermedad lineal de Darier.
Nevo osteovascular anexial porokeratótico (un término
propuesto que incorpora el nevus ostial y dérmico ecrino porokeratótico
[PEODDN] y el nevo folicular poroqueratósico ecrino y folicular del pelo
[PEHFN]) [ 111,112 ]
Además de la poroqueratosis plantaris palmaris y diseminada
(PPPD) y la poroqueratosis punteada, pueden presentarse otros trastornos con
lesiones discretas y queratósicas en las palmas y las plantas de los pies.
Ejemplos incluyen:
Queratosis arsenicales.
Queratodermas palmoplantar
Punteado palmar en el
síndrome de nevo de células basales.
MANEJO
Enfoque general: la
educación sobre la protección solar y la vigilancia clínica para la
transformación maligna son suficientes para el tratamiento de la mayoría de los
pacientes con poroqueratosis. Sin embargo, los pacientes que están preocupados
por la aparición de lesiones o aquellos con enfermedad sintomática pueden
desear una intervención terapéutica.
Las opciones para el tratamiento de la poroqueratosis incluyen
terapia farmacológica tópica, terapias destructivas, escisión quirúrgica y
retinoides orales. Dado que no hay ensayos aleatorios que hayan evaluado las
terapias para la poroqueratosis y los datos disponibles son insuficientes para
las recomendaciones definitivas sobre el tratamiento, la elección de la terapia
se basa principalmente en factores como el tamaño y el número de la lesión, la
ubicación de la lesión, las consideraciones estéticas, la disponibilidad del
tratamiento y la preferencia del paciente. Nuestro enfoque es el siguiente:
Para los pacientes que desean una terapia rápida para
algunas lesiones pequeñas y que no se verán afectados por los cambios
residuales en la pigmentación o cicatrización de la piel, se pueden aplicar
terapias destructivas, como crioterapia, legrado y electrodesecación, terapia
fotodinámica y láser, o escisión quirúrgica.
Para pacientes con lesiones múltiples o grandes, se puede
ofrecer terapia farmacológica tópica con fluorouracilo tópico o imiquimod .
Para pacientes de edad avanzada con lesiones generalizadas, la radioterapia con
rayos Grenz puede ser una opción de tratamiento.
La intensa respuesta inflamatoria que generalmente acompaña
al tratamiento con fluorouracilo tópico o imiquimod limita el área de la piel
que puede tratarse al mismo tiempo. Si se requiere el tratamiento de un área
grande, los retinoides tópicos, como la tretinoína o el tazaroteno , pueden ser
una alternativa, aunque generalmente se requiere una mayor duración del
tratamiento de varios meses.
La respuesta al tratamiento con agentes tópicos a menudo es
impredecible, y los médicos deben permanecer conscientes de que las lesiones
que inicialmente responden bien a cualquier tratamiento pueden reaparecer. Las
opciones adicionales para el tratamiento de la poroqueratosis incluyen
emolientes y queratolíticos tópicos (p. Ej., Ácido salicílico).
Los retinoides sistémicos generalmente se limitan a casos
severos debido a la posibilidad de efectos adversos sistémicos y la alta
probabilidad de recurrencia de la lesión después de la interrupción del
tratamiento.
Protección solar:
debido a la posibilidad de transformación maligna en la poroqueratosis,
se debe alentar a los pacientes a que se protejan de la radiación ultravioleta,
un factor de riesgo conocido para el carcinoma de células escamosas y basales.
Recomendamos ropa de protección solar, protección contra el
sol y uso diario de protección solar de amplio espectro con un factor de
protección solar (FPS) de al menos 30 en áreas de la piel que no pueden
protegerse físicamente. Es de destacar que el uso estricto de protección solar
puede aumentar el riesgo de deficiencia de vitamina D, especialmente en pacientes
con fototipos IV a VI.
Terapias tópicas: no
existen ensayos clínicos aleatorios que evalúen las terapias tópicas de la poroqueratosis,
y su uso se basa en gran medida en la evidencia indirecta de los estudios sobre
el tratamiento de la queratosis actínica (ver "Tratamiento de la
queratosis actínica" ). El régimen / duración del tratamiento óptimo no se
ha determinado, y la respuesta al tratamiento a menudo es impredecible. La
terapia combinada que usa varios agentes tópicos diferentes o agentes tópicos y
terapias destructivas puede ser beneficiosa.
Las opciones terapéuticas tópicas para la poroqueratosis
incluyen:
Fluorouracilo tópico: el fluorouracilo tópico se ha utilizado
en pacientes con poroqueratosis actínica superficial diseminada (DSAP), poroqueratosis
superficial diseminada (DSP), poroqueratosis de Mibelli y poroqueratosis lineal
[ 47,113-116 ]. Se les indica a los pacientes que apliquen una crema tópica de
fluorouracilo al 5% diariamente hasta que se alcance una respuesta inflamatoria
intensa (generalmente varias semanas). La intensa respuesta inflamatoria con
posible ulceración superficial e hiperpigmentación postinflamatoria limita el
uso de fluorouracilo a áreas menos estéticamente importantes.
Imiquimod tópico: el imiquimod tópico se ha utilizado en
informes de casos y pequeñas series de casos de pacientes con porokeratosis de
Mibelli, DSAP, porokeratosis lineal y porokeratosis plantar palmaris y
diseminada (PPPD) [ 117-127 ]. Una revisión sistemática encontró nueve informes
(11 pacientes) de resolución parcial a completa de la porokeratosis de Mibelli
con imiquimod [ 113 ].
Un curso inicial razonable de terapia para imiquimod es la
aplicación de tres a cinco veces por semana durante un período de cuatro a seis
semanas [ 118-120 ]. Se pueden necesitar períodos de tratamiento más largos o
más cortos dependiendo de la respuesta al tratamiento y la aparición de efectos
adversos [ 121,122 ]. Similar al fluorouracilo tópico , se espera una respuesta
inflamatoria local intensa durante el tratamiento con imiquimod.
Retinoides tópicos: la crema de tretinoína al 0.05% y el
gel al 0.1% han sido beneficiosos para el tratamiento de la porokeratosis
lineal [ 128-130 ]. El tazaroteno y el adapaleno también se han utilizado en
algunos pacientes con porokeratosis [ 131 ]. Los retinoides tópicos son menos
irritantes e inducen menos inflamación que el fluorouracilo tópico o imiquimod,
pero requieren un tratamiento más prolongado (10 a 16 semanas o más) [ 128-130
].
Análogos tópicos de la vitamina D: se ha informado que los
análogos tópicos de la vitamina D, como el calcipotriol y el tacalcitol, solo
son efectivos en el DSAP, y pueden requerirse varios meses o más de tratamiento
para mejorar [ 4,132-134 ].
Diclofenaco tópico: el valor del gel tópico de diclofenaco
al 3% no está claro. En una serie de ocho pacientes con DSAP, solo un paciente
tuvo una mejoría parcial después de seis meses de tratamiento [ 135 ]. En un
informe de un solo caso, el diclofenaco tópico fue efectivo para inducir la
resolución parcial de las lesiones inflamatorias de DSAP que persistieron a
pesar del tratamiento con retinoides sistémicos [ 53 ]. Una pequeña serie de
casos sugiere que el medicamento puede ser efectivo para reducir la progresión
de la enfermedad [ 136 ].
Mebutato de ingenol tópico: el mebutato de ingenol, un
agente tópico aprobado para el tratamiento de la queratosis actínica, puede ser
una opción de tratamiento para lesiones de porokeratosis aisladas o limitadas [
137,138 ].
Terapia combinada: según
la experiencia de los autores, el pretratamiento de la piel afectada con
un retinoide tópico, como tretinoína o tazaroteno , durante dos o tres semanas
antes del uso de fluorouracilo tópico o imiquimod parece mejorar la penetración
del fármaco y la respuesta a la terapia. La eficacia relativa de este régimen
para el tratamiento con fluorouracilo tópico o imiquimod solo no se ha
estudiado formalmente en la porokeratosis. Otras combinaciones descritas en
algunos informes de casos incluyen adapaleno con calcipotriol [ 131 ],
imiquimod con fluorouracilo [ 139 ] y crema tópica de fluorouracilo con terapia
fotodinámica (PDT) [ 140]]
Terapias tópicas adyuvantes:
la aplicación de agentes queratolíticos, como el ácido salicílico,
también puede ser beneficiosa [ 141 ]. Se ha informado un tratamiento exitoso
con ácido salicílico en combinación con fluorouracilo o imiquimod [ 126,142 ].
En dos pacientes con porokeratosis de Mibelli, el aclaramiento de la lesión se
logró con cantaridina tópica [ 143 ].
Los corticosteroides tópicos pueden proporcionar alivio
sintomático pero solo una mejora parcial o nula de las lesiones [ 117 ]. Hay un
informe único sobre el uso exitoso de tacrolimus tópico para la poroqueratosis
lineal [ 144 ].
Los emolientes, especialmente con propiedades queratolíticas
(p. Ej., Emolientes que contienen urea), pueden ser útiles en pacientes con
poroqueratosis, ya que pueden reducir la calidad áspera y seca de las lesiones.
Terapias quirúrgicas y destructivas: las lesiones de poroqueratosis
se pueden extirpar o mejorar con procedimientos como la crioterapia, legrado y
electrodesecación, escisión quirúrgica y dermoabrasión [ 145-149 ]. Sin
embargo, todos estos tratamientos están asociados con un alto riesgo de
cicatrización o hiperpigmentación postinflamatoria. Los enfoques alternativos
incluyen:
Terapia con láser: las terapias basadas en la luz también
pueden ser efectivas. El láser de dióxido de carbono se ha utilizado con éxito
para tratar la porokeratosis de Mibelli y la porokeratosis lineal [ 150-153 ].
Se ha informado una mejoría en DSAP o DSP después del tratamiento con un láser
fraccional, láser de rubí de conmutación Q, láser de neodimio: itrio aluminio
granate (Nd: YAG) duplicado en frecuencia y láser de granate erbio: itrio
aluminio (Er: YAG) [ 154 -158 ]. La combinación de láser de dióxido de carbono
y metil aminolevulinato-PDT convencional mostró una mejoría de la condición
pero no una resolución completa de las lesiones DSAP [ 159 ].
Terapia fotodinámica: los informes de casos de PDT
convencional en DSAP y porokeratosis lineal han arrojado resultados variables [
140,160-163 ]. La terapia fotodinámica a la luz del día (DLPDT) ha dado
resultados favorables en cuatro pacientes con DSAP [ 164,165 ].
Radioterapia: en un informe de ocho pacientes con DSAP, la
radioterapia con Grenz y rayos blandos dio como resultado una buena respuesta
en todos los pacientes, con solo una sospecha de recurrencia ( imagen 15 ) [
166 ]. Una reacción inflamatoria e hiperpigmentación postinflamatoria se
produjo en algunos casos y se resolvió espontáneamente en varios meses.
Terapia sistémica: los
retinoides orales, incluidos la acitretina y la isotretinoína , se usan
con poca frecuencia para el tratamiento de casos graves de porokeratosis. Se ha
informado que los retinoides son efectivos en pacientes con DSAP, porokeratosis
lineal, porokeratosis de Mibelli, PPPD y lesiones lineales generalizadas [
69,167-170 ]. Sin embargo, los retinoides sistémicos deben administrarse
durante muchos meses. El grado de mejora es variable; es probable la
recurrencia después de la interrupción del tratamiento. Los retinoides
sistémicos son teratogénicos. La acitretina está contraindicada en pacientes
femeninas en edad fértil; el embarazo debe evitarse durante tres años después
de la interrupción.
En un informe de un solo caso, un paciente con DSAP que
recibió el factor de diferenciación de queratinocitos palifermina para la
profilaxis de mucositis bajo quimioterapia para el tratamiento de tumores
sólidos mostró una mejora notable de DSAP con aclaramiento sostenido de las
lesiones a los 12 meses de seguimiento [ 171 ].
Prevención y vigilancia del cáncer de piel:
aunque la extirpación de lesiones mediante métodos quirúrgicos o
destructivos es una opción para la prevención de la transformación maligna en
las lesiones de porokeratosis, esto generalmente no es necesario ni factible,
especialmente para pacientes con lesiones grandes o extensas. Deben
considerarse factores como el riesgo estimado de malignidad para tipos de
lesiones específicas y el riesgo de defectos cosméticos o funcionales
significativos después de la extracción. La eliminación de las lesiones con el
mayor riesgo de malignidad (porokeratosis lineal o porokeratosis grande de
Mibelli) a menudo daría como resultado una cantidad inaceptable de cicatrices
y, posiblemente, deterioro funcional.
La vigilancia clínica con exámenes periódicos de la piel y
la educación del paciente sobre los signos de advertencia del cáncer de piel y
la protección solar son aspectos clave del tratamiento de todos los pacientes
con pororatosis. Aunque la transformación maligna en DSAP y DSP es rara en
comparación con la porokeratosis de Mibelli y la porokeratosis lineal, la presencia
frecuente de factores de riesgo adicionales de malignidad cutánea (p. Ej., Tipo
de piel clara, antecedentes de alta exposición al sol, inmunosupresión) hace un
seguimiento clínico cercano de estos pacientes valen la pena.
Educación del paciente: todos los pacientes deben recibir
información sobre las señales de advertencia de malignidad. Se debe indicar a
los pacientes que regresen para el seguimiento si se producen cambios en una
lesión previamente estable, como el desarrollo de induración, ulceración,
nodularidad, sangrado, formación de costras o crecimiento rápido. Del mismo
modo, los pacientes deben regresar para la reevaluación si se desarrollan
nuevos síntomas en las lesiones (p. Ej., Sensaciones de hormigueo u hormigueo o
dolor).
Protección solar - Se recomienda la aplicación de las
prácticas de protección solar para reducir el daño actínico.
Seguimiento de rutina: se debe hacer un seguimiento anual de
los pacientes para detectar signos y síntomas de malignidad, detectar cambios
en el estado de salud que puedan sugerir un trastorno sistémico subyacente y
reforzar la educación sobre protección solar. En caso de un número elevado o
lesiones grandes, se deben programar visitas de seguimiento más frecuentes (por
ejemplo, cada seis meses). Cualquier lesión que muestre características
clínicas sospechosas de malignidad (p. Ej., Induración, ulceración, sangrado,
formación de costras, crecimiento rápido) debe extirparse o biopsiarse para un
examen histopatológico.
Si se toma la decisión de extirpar o destruir una lesión con
fines profilácticos, no es necesario hacerlo de manera urgente, ya que el
período entre el desarrollo de la lesión y la neoplasia maligna a menudo abarca
décadas (ver "Transformación maligna" más arriba). Después de la
extracción, el seguimiento clínico aún debe realizarse anualmente para evaluar
a estos pacientes para el desarrollo de lesiones nuevas o recurrentes.
Pacientes inmunocomprometidos: aunque no se ha demostrado que la
inmunosupresión sistémica aumente el riesgo de transformación maligna en las
lesiones de porokeratosis [ 35,97 ], la inmunosupresión es un factor de riesgo
conocido de malignidad cutánea. Por lo tanto, los exámenes de la piel a menudo
se realizan más de una vez al año para la vigilancia del cáncer de piel en esta
población, independientemente de la presencia o ausencia de porokeratosis.
Quimioprevención: los
retinoides sistémicos se usan ocasionalmente para el tratamiento de
pacientes con porokeratosis severa. Según la evidencia indirecta de los
estudios sobre la quimioprevención del carcinoma cutáneo de células escamosas
en receptores de trasplante de órganos sólidos, el riesgo de transformación
maligna puede reducirse durante el tratamiento activo con retinoides sistémicos
[ 172 ]. Sin embargo, esto no se ha estudiado directamente en la porokeratosis,
y se desconoce el impacto a largo plazo de la terapia con retinoides en la
transformación maligna de estas lesiones.
En un ensayo aleatorizado, el uso diario de 1000 mg de
nicotinamida durante 12 meses redujo el número de nuevos carcinomas de células
basales y de células escamosas en un 20 y 30 por ciento, respectivamente, en
pacientes con alto riesgo de cáncer de piel no melanoma [ 173 ]. Sin embargo,
el valor de la suplementación con nicotinamida en la prevención del cáncer de
piel en pacientes con porokeratosis no se ha estudiado específicamente. (Ver
"Epidemiología y factores de riesgo para el carcinoma cutáneo de células
escamosas", sección sobre "Quimioprevención" y "Prevención
y manejo del cáncer de piel en receptores de trasplante de órganos
sólidos", sección sobre "Quimioprevención para SCC" .)
PRONÓSTICO
Sin tratamiento, las lesiones de poroqueratosis generalmente
persisten indefinidamente [ 94 ]. La regresión espontánea es rara [ 35 ].
Se estima que la transformación maligna ocurre en 7 a 11 por
ciento de los pacientes con porokeratosis, con mayor frecuencia en lesiones de
porokeratosis lineal o porokeratosis gigante (una forma de porokeratosis de
Mibelli). La gran mayoría de los carcinomas de células escamosas que se
desarrollan en lesiones de poroqueratosis se tratan con éxito con terapia
local. Sin embargo, se han informado varios casos de carcinoma de células
escamosas metastásicas que surgen en el contexto de porokeratosis [ 97,174-177
].
FUENTE UPTODATE 2020
REFERENCIAS
Kanitakis
J. Porokeratoses: an update of clinical, aetiopathogenic and therapeutic
features. Eur J Dermatol 2014; 24:533.
Williams
GM, Fillman EP. Porokeratosis. In: StatPearls, StatPearls Publishing, Treasure
Island (FL) 2018.
Leow YH, Soon
YH, Tham SN. A report of 31 cases of porokeratosis at the National Skin Centre.
Ann Acad Med Singapore 1996; 25:837.
Niimi Y,
Kawana S. Type 2 segmental manifestation of disseminated superficial actinic
porokeratosis in a 7-year-old girl. Eur J Dermatol 2009; 19:25.
Tan LS,
Chong WS. Porokeratosis in Singapore: an Asian perspective. Australas J
Dermatol 2012; 53:e40.
Gu CY,
Zhang CF, Chen LJ, et al. Clinical analysis and etiology of porokeratosis. Exp
Ther Med 2014; 8:737.
Gracia-Cazaña
T, Vera-Álvarez J, García-Patos V, Gilaberte Y. Imiquimod and Photodynamic
Therapy Are Useful in the Treatment of Porokeratosis in Children with Bone
Marrow Transplantation. Pediatr Dermatol 2015; 32:e291.
Murase J,
Gilliam AC. Disseminated superficial actinic porokeratosis co-existing with
linear and verrucous porokeratosis in an elderly woman: Update on the genetics
and clinical expression of porokeratosis. J Am Acad Dermatol 2010; 63:886.
Xia JH,
Yang YF, Deng H, et al. Identification of a locus for disseminated superficial
actinic porokeratosis at chromosome 12q23.2-24.1. J Invest Dermatol 2000;
114:1071.
Xia K, Deng
H, Xia JH, et al. A novel locus (DSAP2) for disseminated superficial actinic
porokeratosis maps to chromosome 15q25.1-26.1. Br J Dermatol 2002; 147:650.
Liu P,
Zhang S, Yao Q, et al. Identification of a genetic locus for autosomal dominant
disseminated superficial actinic porokeratosis on chromosome 1p31.3-p31.1. Hum
Genet 2008; 123:507.
Luan J, Niu
Z, Zhang J, et al. A novel locus for disseminated superficial actinic
porokeratosis maps to chromosome 16q24.1-24.3. Hum Genet 2011; 129:329.
Cui H, Li
L, Wang W, et al. Exome sequencing identifies SLC17A9 pathogenic gene in two
Chinese pedigrees with disseminated superficial actinic porokeratosis. J Med
Genet 2014; 51:699.
Leng Y, Yan
L, Feng H, et al. Mutations in mevalonate pathway genes in patients with
familial or sporadic porokeratosis. J Dermatol 2018; 45:862.
Zhang SQ,
Jiang T, Li M, et al. Exome sequencing identifies MVK mutations in disseminated
superficial actinic porokeratosis. Nat Genet 2012; 44:1156.
Zhou Y, Liu J, Fu X, et al. Identification of three novel frameshift mutations
of the MVK gene in four Chinese families with disseminated superficial actinic
porokeratosis. Br J Dermatol 2013; 169:193.
Li M, Li Z,
Wang J, et al. Mutations in the mevalonate pathway genes in Chinese patients
with porokeratosis. J Eur Acad Dermatol Venereol 2016; 30:1512.
Zhang Z, Li
C, Wu F, et al. Genomic variations of the mevalonate pathway in porokeratosis.
Elife 2015; 4:e06322.
Moir RD,
Spann TP. The structure and function of nuclear lamins: implications for
disease. Cell Mol Life Sci 2001; 58:1748.
Jin R, Luo
X, Luan K, et al. Disorder of the mevalonate pathway inhibits calcium-induced
differentiation of keratinocytes. Mol Med Rep 2017; 16:4811.
Wei S, Yang
S, Lin D, et al. A novel locus for disseminated superficial porokeratosis maps
to chromosome 18p11.3. J Invest Dermatol 2004; 123:872.
Wei S,
Zhang TD, Zhou Y, et al. Fine mapping of the disseminated superficial
porokeratosis locus to a 2.7 Mb region at 18p11.3. Clin Exp Dermatol 2010;
35:664.
Wei SC,
Yang S, Li M, et al. Identification of a locus for porokeratosis palmaris et
plantaris disseminata to a 6.9-cM region at chromosome 12q24.1-24.2. Br J
Dermatol 2003; 149:261.
Happle R.
Mibelli revisited: a case of type 2 segmental porokeratosis from 1893. J Am
Acad Dermatol 2010; 62:136.
Chernosky
ME, Freeman RG. Disseminated superficial actinic porokeratosis (DSAP). Arch
Dermatol 1967; 96:611.
Neumann RA,
Knobler RM, Jurecka W, Gebhart W. Disseminated superficial actinic
porokeratosis: experimental induction and exacerbation of skin lesions. J Am
Acad Dermatol 1989; 21:1182.
Shin EJ,
Gwak MJ, Jeong KH, Lee MH. Disseminated Superficial Actinic Porokeratosis in a
Vitiligo Patient Undergoing Treatment with Long-Term Narrowband Ultraviolet B.
Ann Dermatol 2018; 30:249.
Zhao MJ,
Abdul-Fattah B, Qu XY, et al. Mycosis fungoides patient accompanied actinic
keratosis, actinic keratosis with squamous cell carcinoma transformation, and
porokeratosis after NBUVB therapy - 1st case report and review of the
literature. Medicine (Baltimore) 2016; 95:e5043.
Schwarz T,
Seiser A, Gschnait F. Disseminated superficial "actinic"
porokeratosis. J Am Acad Dermatol 1984; 11:724.
Raychaudhuri
SP, Smoller BR. Porokeratosis in immunosuppressed and nonimmunosuppressed
patients. Int J Dermatol 1992; 31:781.
Rodríguez EA, Jakubowicz S, Chinchilla DA, et al. Porokeratosis of Mibelli and
HIV-infection. Int J Dermatol 1996; 35:402.
Herranz P, Pizarro A, De Lucas R, et al. High incidence of porokeratosis in renal
transplant recipients. Br J Dermatol 1997; 136:176.
Alexis AF,
Busam K, Myskowski PL. Porokeratosis of Mibelli following bone marrow
transplantation. Int J Dermatol 2006; 45:361.
Komorowski
RA, Clowry LJ. Porokeratosis of mibelli in transplant recipients. Am J Clin
Pathol 1989; 91:71.
Kanitakis
J, Euvrard S, Faure M, Claudy A. Porokeratosis and immunosuppression. Eur J
Dermatol 1998; 8:459.
Rothman IL,
Wirth PB, Klaus MV. Porokeratosis of Mibelli following heart transplant. Int J
Dermatol 1992; 31:52.
Bednarek R,
Ezra N, Toubin Y, et al. Eruptive disseminated porokeratosis associated with
corticosteroid-induced immunosuppression. Clin Exp Dermatol 2015; 40:753.
Sertznig P,
von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol
Venereol 2012; 26:404.
Shoimer I,
Robertson LH, Storwick G, Haber RM. Eruptive disseminated porokeratosis: a new
classification system. J Am Acad Dermatol 2014; 71:398.
Kanitakis
J, Misery L, Nicolas JF, et al. Disseminated superficial porokeratosis in a
patient with AIDS. Br J Dermatol 1994; 131:284.
Gilead L,
Guberman D, Zlotogorski A, et al. Immunosuppression-induced porokeratosis of
Mibelli: Complete regression of lesions upon cessation of immunosuppressive
therapy. J Eur Acad Dermatol Venereol 1995; 5:170.
Tsambaos D,
Spiliopoulos T. Disseminated superficial porokeratosis: complete remission
subsequent to discontinuation of immunosuppression. J Am Acad Dermatol 1993;
28:651.
Yazkan F,
Turk BG, Dereli T, Kazandi AC. Porokeratosis of Mibelli induced by topical
corticosteroid. J Cutan Pathol 2006; 33:516.
James AJ,
Clarke LE, Elenitsas R, Katz K. Segmental porokeratosis after radiation therapy
for follicular lymphoma. J Am Acad Dermatol 2008; 58:S49.
Romaní J,
Pujol RM, Casanova JM, de Moragas JM. Disseminated superficial porokeratosis
developing after electron-beam total skin irradiation for mycosis fungoides.
Clin Exp Dermatol 1996; 21:310.
Nova MP,
Goldberg LJ, Mattison T, Halperin A. Porokeratosis arising in a burn scar. J Am
Acad Dermatol 1991; 25:354.
Dippel E,
Haas N, Czarnetzki BM. Porokeratosis of Mibelli associated with active chronic
hepatitis and vitiligo. Acta Derm Venereol 1994; 74:463.
Hunt SJ,
Sharra WG, Abell E. Linear and punctate porokeratosis associated with end-stage
liver disease. J Am Acad Dermatol 1991; 25:937.
Cannavó SP,
Borgia F, Adamo B, Guarneri B. Simultaneous development and parallel course of
disseminated superficial porokeratosis and ovarian cancer: Coincidental
association or true paraneoplastic syndrome? J Am Acad Dermatol 2008; 58:657.
Kono T,
Kobayashi H, Ishii M, et al. Synchronous development of disseminated
superficial porokeratosis and hepatitis C virus-related hepatocellular
carcinoma. J Am Acad Dermatol 2000; 43:966.
Morton CA,
Shuttleworth D, Douglas WS. Porokeratosis and Crohn's disease. J Am Acad
Dermatol 1995; 32:894.
Sawyer R,
Picou KA. Facial presentation of disseminated superficial actinic
porokeratosis. Ear Nose Throat J 1989; 68:57.
Shimizu S,
Takashima Y, Hotta M, et al. Inflammatory disseminated superficial
porokeratosis successfully controlled with a combination of topical diclofenac
gel and systemic etretinate. J Eur Acad Dermatol Venereol 2018; 32:e201.
Bencini PL, Tarantino A, Grimalt R, et al. Porokeratosis and immunosuppression. Br J
Dermatol 1995; 132:74.
Chun SI, Lee JS, Kim NS, Park KD. Disseminated epidermolytic acanthoma with
disseminated superficial porokeratosis and verruca vulgaris in an
immunosuppressed patient. J Dermatol 1995; 22:690.
Rio B, Magana C, Le Tourneau A, et al. Disseminated superficial porokeratosis after
autologous bone marrow transplantation. Bone Marrow Transplant 1997; 19:77.
Knoell KA,
Patterson JW, Wilson BB. Sudden onset of disseminated porokeratosis of Mibelli
in a renal transplant patient. J Am Acad Dermatol 1999; 41:830.
Bozdağ KE,
Biçakçi H, Ermete M. Giant porokeratosis. Int J Dermatol 2004; 43:518.
Raychaudhury
T, Valsamma DP. Giant porokeratosis. Indian J Dermatol Venereol Leprol 2011;
77:601.
Robinson
JB, Im DD, Jockle G, Rosenshein NB. Vulvar porokeratosis: case report and
review of the literature. Int J Gynecol Pathol 1999; 18:169.
Neri I,
Marzaduri S, Passarini B, Patrizi A. Genital porokeratosis of Mibelli.
Genitourin Med 1995; 71:410.
Koley S,
Sarkar J, Choudhary S, et al. Different morphological variants of hypertrophic
porokeratosis and disseminated lesions of porokeratosis of Mibelli: a rare
co-existence. Indian J Dermatol Venereol Leprol 2011; 77:199.
Rajesh G,
Devan P, Keerthi S, Karthikeyan K. Acral porokeratosis associated with
anonychia. Indian J Dermatol Venereol Leprol 2018; 84:81.
Thomas C,
Ogboli MI, Carr RA, Charles-Holmes R. Hypertrophic perianal porokeratosis in
association with superficial actinic porokeratosis of the leg. Clin Exp
Dermatol 2003; 28:676.
Friedman B,
Golubets K, Ho J, Patton T. Linear porokeratosis associated with multiple
squamous cell carcinomas. Cutis 2017; 100:E11.
Sommerlad
M, Lock A, Moir G, et al. Linear porokeratosis with multiple squamous cell carcinomas
successfully treated by electrochemotherapy. Br J Dermatol 2016;
175:1342.
Guenova E, Hoetzenecker W, Metzler G, et al. Multicentric Bowen disease in linear
porokeratosis. Eur J Dermatol 2007; 17:439.
Happle R.
Cancer proneness of linear porokeratosis may be explained by allelic loss.
Dermatology 1997; 195:20.
Hartman R,
Mandal R, Sanchez M, Stein JA. Porokeratosis plantaris, palmaris, et
disseminata. Dermatol Online J 2010; 16:22.
Marschalkó
M, Somlai B. Porokeratosis plantaris, palmaris, et disseminata. Arch Dermatol
1986; 122:890.
Irisawa R,
Yamazaki M, Yamamoto T, Tsuboi R. A case of porokeratosis plantaris palmaris et
disseminata and literature review. Dermatol Online J 2012; 18:5.
Shaw JC,
White CR Jr. Porokeratosis plantaris palmaris et disseminata. J Am Acad
Dermatol 1984; 11:454.
Patrizi A,
Passarini B, Minghetti G, Masina M. Porokeratosis palmaris et plantaris
disseminata: an unusual clinical presentation. J Am Acad Dermatol 1989; 21:415.
Lestringant
GG, Berge T. Porokeratosis punctata palmaris et plantaris. A new entity? Arch
Dermatol 1989; 125:816.
Sakas EL,
Gentry RH. Porokeratosis punctata palmaris et plantaris (punctate
porokeratosis). Case report and literature review. J Am Acad Dermatol 1985;
13:908.
Tallon B,
Blumental G, Bhawan J. Porokeratosis ptychotropica: a lesser-known variant.
Clin Exp Dermatol 2009; 34:e895.
McGuigan K,
Shurman D, Campanelli C, Lee JB. Porokeratosis ptychotropica: a clinically
distinct variant of porokeratosis. J Am Acad Dermatol 2009; 60:501.
Yeo J,
Winhoven S, Tallon B. Porokeratosis ptychotropica: a rare and evolving variant
of porokeratosis. J Cutan Pathol 2013; 40:1042.
D'souza P,
Dhali TK, Arora S, et al. Porokeratosis ptychotropica: a rare variant of
porokeratosis. Dermatol Online J 2014; 20.
Wanat KA,
Gormley RH, Bennett DD, Kovarik CL. Genitogluteal porokeratosis involving the
scrotum: an unusual presentation of an uncommon disease. J Cutan Pathol 2012;
39:72.
Joshi R,
Minni K. Genitogluteal porokeratosis: a clinical review. Clin Cosmet Investig
Dermatol 2018; 11:219.
Joshi R,
Jadhav Y. Penoscrotal porokeratosis: A distinct entity. Indian Dermatol Online
J 2015; 6:339.
Liang C, Batra P, Patel R, Kamino H. Genital porokeratosis. Dermatol Online J 2009; 15:23.
Lee Y, Choi
EH. Exclusive facial porokeratosis: histopathologically showing follicular
cornoid lamellae. J Dermatol 2011; 38:1072.
Rocha-Sousa VL, Costa JB, de Aquino Paulo-Filho T, et al. Follicular porokeratosis on the
face. Am J Dermatopathol 2011; 33:636.
Wang NS,
Gruson LM, Kamino H. Facial follicular porokeratosis: a case report. Am J
Dermatopathol 2010; 32:720.
Kim J, Wood
BA, Harvey NT. Follicular porokeratosis of the nose: two further cases of an
emerging variant of porokeratosis. Pathology 2015; 47:482.
Kanekura T,
Yoshii N. Eruptive pruritic papular porokeratosis: a pruritic variant of
porokeratosis. J Dermatol 2006; 33:813.
Kanzaki T,
Miwa N, Kobayashi T, Ogawa S. Eruptive pruritic papular porokeratosis. J
Dermatol 1992; 19:109.
Stork J,
Kodetová D. Disseminated superficial porokeratosis: an eruptive pruritic
papular variant. Dermatology 1997; 195:304.
Kaushik A,
Handa S, Chatterjee D, et al. Disseminated filiform hyperkeratosis - a variant
of porokeratosis? J Eur Acad Dermatol Venereol 2018; 32:e419.
Flanagan N,
Boyadjiev SA, Harper J, et al. Familial craniosynostosis, anal anomalies, and
porokeratosis: CAP syndrome. J Med Genet 1998; 35:763.
Mendoza-Londono
R, Lammer E, Watson R, et al. Characterization of a new syndrome that
associates craniosynostosis, delayed fontanel closure, parietal foramina,
imperforate anus, and skin eruption: CDAGS. Am J Hum Genet 2005; 77:161.
Sasson M,
Krain AD. Porokeratosis and cutaneous malignancy. A review. Dermatol Surg 1996;
22:339.
Maubec E,
Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J
Dermatol 2005; 152:1389.
Goerttler
EA, Jung EG. Porokeratosis [correction of Parakeratosis] Mibelli and skin
carcinoma: a critical review. Humangenetik 1975; 26:291.
Silver SG,
Crawford RI. Fatal squamous cell carcinoma arising from transplant-associated
porokeratosis. J Am Acad Dermatol 2003; 49:931.
Otsuka F,
Umebayashi Y, Watanabe S, et al. Porokeratosis large skin lesions are
susceptible to skin cancer development: histological and cytological
explanation for the susceptibility. J Cancer Res Clin Oncol 1993; 119:395.
Lin JH, Hsu
MM, Sheu HM, Lee JY. Coexistence of three variants of porokeratosis with
multiple squamous cell carcinomas arising from lesions of giant hyperkeratotic
porokeratosis. J Eur Acad Dermatol Venereol 2006; 20:621.
Uenishi T, Teramura K, Kitamura M, et al. Hyperkeratotic variant of porokeratosis Mibelli
with dermal amyloid deposits. J Dermatol 2010; 37:475.
Carlesimo M, Rossi A, Fidanza L, et al. Disseminated Superficial Porokeratosis with
Dermal Amyloid Deposits. Case Rep Dermatol 2009; 1:35.
Tan TS,
Tallon B. Pigmented Porokeratosis. A Further Variant? Am J Dermatopathol 2016;
38:218.
Wade TR,
Ackerman AB. Cornoid lamellation. A histologic reaction pattern. Am J
Dermatopathol 1980; 2:5.
Zaballos P,
Puig S, Malvehy J. Dermoscopy of disseminated superficial actinic
porokeratosis. Arch Dermatol 2004; 140:1410.
Panasiti V,
Rossi M, Curzio M, et al. Disseminated superficial actinic porokeratosis
diagnosed by dermoscopy. Int J Dermatol 2008; 47:308.
Uhara H,
Kamijo F, Okuyama R, Saida T. Open pores with plugs in porokeratosis clearly
visualized with the dermoscopic furrow ink test: report of 3 cases. Arch
Dermatol 2011; 147:866.
Oiso N,
Kawada A. Dermoscopic features in disseminated superficial actinic
porokeratosis. Eur J Dermatol 2011; 21:439.
Pizzichetta
MA, Canzonieri V, Massone C, Soyer HP. Clinical and dermoscopic features of
porokeratosis of Mibelli. Arch Dermatol 2009; 145:91.
Urbina F,
Pérez A, Requena L, Rütten A. Circumscribed palmar or plantar hypokeratosis 10
years after the first description: what is known and the issues under
discussion. Actas Dermosifiliogr 2014; 105:574.
Elfatoiki
FZ, Soussi W, Chiheb S, et al. Cutaneous sarcoidosis simulating porokeratosis
of Mibelli. Pan Afr Med J 2015; 20:195.
Llamas-Velasco
M, Hilty N, Kempf W. Porokeratotic adnexal ostial naevus: review on the entity
and therapeutic approach. J Eur Acad Dermatol Venereol 2015; 29:2032.
Goddard DS,
Rogers M, Frieden IJ, et al. Widespread porokeratotic adnexal ostial nevus:
clinical features and proposal of a new name unifying porokeratotic eccrine
ostial and dermal duct nevus and porokeratotic eccrine and hair follicle nevus.
J Am Acad Dermatol 2009; 61:1060.e1.
Shelley WB,
Shelley ED. Disseminated superficial porokeratosis: rapid therapeutic response
to 5-fluorouracil. Cutis 1983; 32:139.
Gonçalves
JC. Fluorouracil ointment treatment of porokeratosis of Mibelli. Arch Dermatol
1973; 108:131.
McDonald
SG, Peterka ES. Porokeratosis (Mibelli): treatment with topical 5-fluorouracil.
J Am Acad Dermatol 1983; 8:107.
Balato N,
Di Nardo G, Boccia L, Nappa P. [Linear Mibelli's porokeratosis. Treatment with
topical 5-fluorouracil]. G Ital Dermatol Venereol 1986; 121:147.
Weidner T,
Illing T, Miguel D, Elsner P. Treatment of Porokeratosis: A Systematic Review.
Am J Clin Dermatol 2017; 18:435.
Harrison S,
Sinclair R. Porokeratosis of Mibelli: successful treatment with topical 5%
imiquimod cream. Australas J Dermatol 2003; 44:281.
Montes-De-Oca-Sánchez
G, Tirado-Sánchez A, García-Ramírez V. Porokeratosis of Mibelli of the axillae:
treatment with topical imiquimod. J Dermatolog Treat 2006; 17:319.
Arun B,
Pearson J, Chalmers R. Disseminated superficial actinic porokeratosis treated
effectively with topical imiquimod 5% cream. Clin Exp Dermatol 2011; 36:509.
Jain S.
Successful treatment of porokeratosis of Mibelli with imiquimod 5% cream. Clin
Exp Dermatol 2006; 31:302.
Ahn SJ, Lee
HJ, Chang SE, et al. Case of linear porokeratosis: successful treatment with
topical 5% imiquimod cream. J Dermatol 2007; 34:146.
Agarwal S,
Berth-Jones J. Porokeratosis of Mibelli: successful treatment with 5% imiquimod
cream. Br J Dermatol 2002; 146:338.
Jensen JM,
Egberts F, Proksch E, Hauschild A. Disseminated porokeratosis palmaris and
plantaris treated with imiquimod cream to prevent malignancy. Acta Derm
Venereol 2005; 85:550.
Esser AC,
Pittelkow MR, Randle HW. Human papillomavirus isolated from
transplant-associated porokeratoses of mibelli responsive to topical 5%
imiquimod cream. Dermatol Surg 2006; 32:858.
Erbagci Z,
Tuncel AA, Erbagci I. Successful treatment of porokeratosis with topical
imiquimod in 2 immunosuppressed cases. J Drugs Dermatol 2006; 5:668.
Gajic B,
Tang K, Whitfeld M. Porokeratosis of Mibelli: Involution and resolution with 5%
imiquimod cream. Australas J Dermatol 2011; 52:301.
Grover C,
Goel A, Nanda S, et al. A case of extensive linear porokeratosis with
evaluation of topical tretinoin versus 5-flourouracil as treatment modalities.
J Dermatol 2005; 32:1000.
Dervis E,
Demirkesen C. Generalized linear porokeratosis. Int J Dermatol 2006; 45:1077.
Agrawal SK,
Gandhi V, Madan V, Bhattacharya SN. Topical tretinoin in Indian male with
zosteriform porokeratosis. Int J Dermatol 2003; 42:919.
Nakamura Y,
Yamaguchi M, Nakamura A, Muto M. Calcipotriol and adapalene therapy for
disseminated superficial actinic porokeratosis. Indian J Dermatol Venereol
Leprol 2014; 80:373.
Böhm M,
Luger TA, Bonsmann G. Disseminated superficial actinic porokeratosis: treatment
with topical tacalcitol. J Am Acad Dermatol 1999; 40:479.
Abe M,
Yokoyama Y, Ishikawa O. Successful treatment of disseminated superficial
actinic porokeratosis with tacalcitol lotion. J Dermatol 2010; 37:913.
Harrison
PV, Stollery N. Disseminated superficial actinic porokeratosis responding to
calcipotriol. Clin Exp Dermatol 1994; 19:95.
Vlachou C,
Kanelleas AI, Martin-Clavijo A, Berth-Jones J. Treatment of disseminated
superficial actinic porokeratosis with topical diclofenac gel: a case series. J
Eur Acad Dermatol Venereol 2008; 22:1343.
Marks S,
Varma R, Cantrell W, et al. Diclofenac sodium 3% gel as a potential treatment
for disseminated superficial actinic porokeratosis. J Eur Acad Dermatol
Venereol 2009; 23:42.
Kindem S, Serra-Guillén C, Sorní G, et al. Treatment of porokeratosis of Mibelli with
ingenol mebutate: a possible new therapeutic option. JAMA Dermatol 2015;
151:85.
Anderson I,
Routt ET, Jim On SC. Disseminated superficial actinic porokeratosis treated
with ingenol mebutate gel 0.05. Cutis 2017; 99:E36.
Venkatarajan S, LeLeux TM, Yang D, et al. Porokeratosis of Mibelli: Successful treatment
with 5 percent topical imiquimod and topical 5 percent 5-fluorouracil. Dermatol
Online J 2010; 16:10.
Levitt J,
Emer JJ, Emanuel PO. Treatment of porokeratosis of mibelli with combined use of
photodynamic therapy and Fluorouracil cream. Arch Dermatol 2010; 146:371.
Jih MH.
Porokeratosis plantaris, palmaris, et disseminata. Dermatol Online J 2003;
9:34.
Danby W.
Treatment of porokeratosis with fluorouracil and salicylic acid under
occlusion. Dermatol Online J 2003; 9:33.
Levitt JO,
Keeley BR, Phelps RG. Treatment of porokeratosis of Mibelli with cantharidin. J
Am Acad Dermatol 2013; 69:e254.
Parks AC,
Conner KJ, Armstrong CA. Long-term clearance of linear porokeratosis with
tacrolimus, 0.1%, ointment. JAMA Dermatol 2014; 150:194.
Bhushan M,
Craven NM, Beck MH, Chalmers RJ. Linear porokeratosis of mibelli: successful
treatment with cryotherapy. Br J Dermatol 1999; 141:389.
Chowdhury
MM, Inaloz HS, Holt PJ. A scaly macule on the bridge of the nose of a
15-year-old boy. Pediatr Dermatol 2000; 17:149.
Carranza DC, Haley JC, Chiu M. Facial porokeratosis. Skinmed 2008; 7:51.
Cohen PR,
Held JL, Katz BE. Linear porokeratosis: successful treatment with diamond
fraise dermabrasion. J Am Acad Dermatol 1990; 23:975.
Spencer JM,
Katz BE. Successful treatment of porokeratosis of Mibelli with diamond fraise
dermabrasion. Arch Dermatol 1992; 128:1187.
Barnett JH.
Linear porokeratosis: treatment with the carbon dioxide laser. J Am Acad
Dermatol 1986; 14:902.
Rabbin PE,
Baldwin HE. Treatment of porokeratosis of Mibelli with CO2 laser vaporization
versus surgical excision with split-thickness skin graft. A comparison. J
Dermatol Surg Oncol 1993; 19:199.
Hunziker T,
Bayard W. Carbon dioxide laser in the treatment of porokeratosis. J Am Acad
Dermatol 1987; 16:625.
McCullough
TL, Lesher JL Jr. Porokeratosis of Mibelli: rapid recurrence of a large lesion
after carbon dioxide laser treatment. Pediatr Dermatol 1994; 11:267.
Chrastil B,
Glaich AS, Goldberg LH, Friedman PM. Fractional photothermolysis: a novel
treatment for disseminated superficial actinic porokeratosis. Arch Dermatol
2007; 143:1450.
Itoh M,
Nakagawa H. Successful treatment of disseminated superficial actinic
porokeratosis with Q-switched ruby laser. J Dermatol 2007; 34:816.
Lolis MS,
Marmur ES. Treatment of disseminated superficial actinic porokeratosis (DSAP)
with the Q-switched ruby laser. J Cosmet Laser Ther 2008; 10:124.
Liu HT.
Treatment of lichen amyloidosis (LA) and disseminated superficial porokeratosis
(DSP) with frequency-doubled Q-switched Nd:YAG laser. Dermatol Surg 2000;
26:958.
Rosenblum
J, Roenigk HH Jr. Erbium laser for the treatment of disseminated superficial
actinic porokeratosis: a case report. Dermatol Surg 2013; 39:1543.
Kim HS,
Baek JH, Park YM, et al. Photodynamic Therapy Combined with CO(2) Laser
Vaporization on Disseminated Superficial Actinic Porokeratosis: A Report of 2
Cases on the Face. Ann Dermatol 2011; 23:S211.
Nayeemuddin
FA, Wong M, Yell J, Rhodes LE. Topical photodynamic therapy in disseminated
superficial actinic porokeratosis. Clin Exp Dermatol 2002; 27:703.
Fernández-Guarino M, Harto A, Pérez-Garcia B, et al. Photodynamic therapy in disseminated
superficial actinic porokeratosis. J Eur Acad Dermatol Venereol 2009; 23:176.
Cavicchini S,
Tourlaki A. Successful treatment of disseminated superficial actinic
porokeratosis with methyl aminolevulinate-photodynamic therapy. J Dermatolog
Treat 2006; 17:190.
García-Navarro
X, Garcés JR, Baselga E, Alomar A. Linear porokeratosis: excellent response to
photodynamic therapy. Arch Dermatol 2009; 145:526.
Salas T, Hernandez-Gil J, Lopez A, et al. Two cases of disseminated superficial actinic
porokeratosis treated with daylight-mediated photodynamic therapy. Dermatol
Ther 2016; 29:484.
Ferrer
Guillén B, Giácaman MM, Pérez Ferriols A. Improved effect on 2 cases of
disseminated superficial actinic porokeratosis with daylight photodynamic
therapy. Photodiagnosis Photodyn Ther 2018; 23:365.
Ramelyte E,
Bylaite-Bucinskiene M, Dummer R, Imhof L. Successful Use of Grenz Rays for
Disseminated Superficial Actinic Porokeratosis: Report of 8 Cases. Dermatology
2017; 233:217.
McCallister
RE, Estes SA, Yarbrough CL. Porokeratosis plantaris, palmaris, et disseminata.
Report of a case and treatment with isotretinoin. J Am Acad Dermatol 1985;
13:598.
Hong JB,
Hsiao CH, Chu CY. Systematized linear porokeratosis: a rare variant of diffuse
porokeratosis with good response to systemic acitretin. J Am Acad Dermatol
2009; 60:713.
Garg T,
Ramchander, Varghese B, et al. Generalized linear porokeratosis: a rare entity
with excellent response to acitretin. Dermatol Online J 2011; 17:3.
Gutierrez EL, Galarza C, Ramos W, et al. Facial porokeratosis: A series of six patients.
Australas J Dermatol 2010; 51:191.
Howard M,
Hall A. A report and follow up of a patient with disseminated superficial
actinic porokeratosis (DSAP) undergoing novel systemic treatment with
palifermin (a keratinocyte growth factor) during chemotherapy. Dermatol Online
J 2018; 24.
Chen K,
Craig JC, Shumack S. Oral retinoids for the prevention of skin cancers in solid
organ transplant recipients: a systematic review of randomized controlled
trials. Br J Dermatol 2005; 152:518.
Chen AC,
Martin AJ, Choy B, et al. A Phase 3 Randomized Trial of Nicotinamide for
Skin-Cancer Chemoprevention. N Engl J Med 2015; 373:1618.
Rongioletti
F, Rebora A. Disseminated porokeratosis with fatal metastatic squamous cell
carcinoma: an additional case of "malignant disseminated
porokeratosis". Am J Dermatopathol 2002; 24:144.
Sawai T,
Hayakawa H, Danno K, et al. Squamous cell carcinoma arising from giant
porokeratosis: a case with extensive metastasis and hypercalcemia. J Am Acad
Dermatol 1996; 34:507.
Lozinski
AZ, Fisher BK, Walter JB, Fitzpatrick PJ. Metastatic squamous cell carcinoma in
linear porokeratosis of Mibelli. J Am Acad Dermatol 1987; 16:448.
Anzai S,
Takeo N, Yamaguchi T, et al. Squamous cell carcinoma in a renal transplant
recipient with linear porokeratosis. J Dermatol 1999; 26:244.