Un paciente de 34 años con fibrosis quística se internó en una unidad de
cuidados respiratorios por presentar fiebre, dolor y distensión abdominal.
El paciente estaba en lista de espera para transplante pulmonar por
insuficiencia respiratoria secundaria a fibrosis quística. La fibrosis quística
había sido diagnosticada en el nacimiento, y era homocigoto para la mutación
F508 del gen regulador de la proteína de conductancia de transmembrana (CFTR).
Había sido internado al menos una vez por año por complicaciones de su
enfermedad, la última vez hacía un año por neumonía causada por Pseudomona
aeruginosa y Staphylococcus aureus meticilino resistente.
Su tratamiento respiratorio incluía nebulizaciones con albuterol y solución
salina hipertónica, seguidos secuencialmente de terapia física con un sistema
de oscilación de tórax (High Frequency Chest Wall Oscillation) (HFCWO), y
nebulizaciones de colistin. Otras medicaciones incluían
trimetoprima-sulfametoxazol dos veces por día; durante los dos meses antes de la
internación, se le administraron aerosoles de ciprofloxacina y tobramicina por
empeoramiento de la tos, y deterioro de la función pulmonar. En los cultivos de
esputo había desarrollado P. aeruginosa.
Tres días antes de la internación, el paciente había sido visto por
consultorio externo por tos, aumento de la disnea, y aumento moderado de la
producción de esputo.
En el examen el paciente estaba disneico y se auscultaban rales
inspiratorios en ambos campos pulmonares superiores. La saturación de oxígeno
era de 93% respirando aire ambiente. El volumen espiratorio forzado en 1
segundo (VEF1) era de 0,74 litros (21% del predicho). De una muestra de esputo
obtenida desarrolló P. aeruginosa con resistencia intermedia a
fluoroquinolonas; era susceptible a cefepime, ceftazidima, aztreonam,
gentamicina, y piperacilina-tazobactam. Fue entonces internado para soporte
respiratorio y antibióticoterapia adicional.
Una cirugía intestinal había sido llevada a cabo cuando era muy pequeño.
Era diabético insulino-dependiente y tenía osteoporosis. Aproximadamente 3,5
años antes de la internación actual, durante una evaluación para transplante de
pulmón, se observó un aumento leve de transaminasas por lo que se llevó a cabo
una biopsia hepática que mostró tejido cicatrizal en el árbol biliar e
inflamación, leve fibrosis sin cirrosis y leve a moderada esteatosis.
Diecinueve meses antes de la internación actual presentó un cuadro de
obstrucción intestinal que fue tratado con líquidos intravenosos y
polietilenglicol.
El paciente tenía psoriasis, mal estado dentario, y fractura de muñeca post
traumática. No presentaba alergias conocidas. Era soltero y vivía con su padre
y su madrastra que tenía como mascota un canario. Trabajaba en la industria de
alimentos, raramente consumía alcohol, y había fumado por un período breve de
tiempo 15 años atrás. No consumía drogas
ilícitas. No había estado expuesto a tuberculosis o personas enfermas ni había
realizado viajes recientemente. No había antecedentes familiares de fibrosis
quística, enfermedades cardíacas, diabetes mellitus, o cáncer.
Su madre había fallecido en edad joven de causa desconocida. Los
medicamentos que consumía consistían en solución salina hipertónica en
nebulizaciones, ursodiol, trimetoprima-sulfametoxazol, pancreolipasa, insulina
NPH con las comidas e insulina lispro según necesidad, alendronato
semanalmente, y vitaminas.
En el examen el paciente estaba aparentemente bien nutrido pero era delgado
y pesaba 54 kg. Era capaz de conversar sin disnea o taquipnea. La presión sanguínea
era de 138/57 mm Hg, el pulso 73 por minuto, la temperatura de 36,2°C, y la
frecuencia respiratoria de 18 por minuto; la saturación de oxígeno 91%
respirando aire ambiente, y 97% respirando 2 litros de oxígeno por cánula
nasal.
La excursión del tórax era simétrica y era 2 a 3 cm; había sonidos
respiratorios gruesos bilateralmente con rales inspiratorios en los campos
medios y superiores en región posterior del pulmón izquierdo, y campo superior
y posterior del derecho, y en ambas regiones superiores de la cara anterior del
tórax.
El abdomen era blando, sin zonas dolorosas ni distensión. Los sonidos
intestinales estaban presentes. Había una cicatriz quirúrgica transversa de 15
cm de largo en el abdomen, y el reborde hepático se palpaba 1 cm por debajo del
margen costal; no había dolor ni signo de rebote o irritación peritoneal.
Tenía hipocratismo en los dedos de manos y pies; el resto del examen era
normal. El tiempo de protrombina era de 16,9 segundos (normal de 11,1 a 13,6);
el resto de los tests de coagulación, nivel plasmático de bilirrubina,
proteínas totales, albúmina, globulina, magnesio, amilasa, y LDH, eran
normales; otros tests de laboratorio se muestran en la Tabla 1. Una Rx de tórax
mostró pulmones hiperexpandidos con distorsión de la arquitectura consistente
con fibrosis quística; los hallazgos de la radiografía no mostraban cambios
respecto de estudios previos.

Tabla 1. Datos de laboratorio.
En la columna de la izquierda se muestran los valores normales para un
adulto. En las otras tres columnas de izquierda a derecha se muestran los datos
de laboratorio del día del ingreso, del 8° y 9° días de internación
respectivamente.
En la tarde del quinto día de internación, la temperatura subió a 38,7°C;
el examen pulmonar y abdominal no mostraba cambios. Se obtuvieron muestras de
sangre, esputo, y orina para cultivo. Una radiografía de tórax mostró el
catéter extendiéndose desde el brazo derecho con la punta proyectándose sobre
el hombro derecho, no mostrando cambios respecto al control realizado durante
su colocación. Se administró acetaminofén y se continuó con los antibióticos.
La mañana siguiente la temperatura era de 36,8 |C pero ascendió a 38,7°C más
tarde ese mismo día. Tenía tos con expectoración de un esputo verde con estrías
amarronadas. Había roncus inspiratorios en la base izquierda y en ambos
vértices, y se escuchaban rales en vértices. Los ruidos intestinales eran
normales, y no había dolor abdominal ni distensión. Los cultivos de sangre y
orina fueron negativos y el esputo permaneció sin cambios. Se sacó el catéter y
se mandó a cultivo la punta del mismo. Se comenzó con vancomicina.
En el octavo día de hospital la temperatura aumentó a 39,1°C y comenzó con
náuseas y vómitos. En el examen la presión sanguínea era de 99/52 mm Hg, el
pulso de 117 por minuto, la frecuencia respiratoria de 18 por minuto, y la
saturación de oxígeno de 90% respirando aire ambiente. El paciente impresionaba
estar bien. Había rales en los campos superiores con algunas sibilancias. Los
sonidos intestinales estaban normales; el cuadrante superior derecho del
abdomen dolía, y no había distensión ni rebote. El resultado de los tests de
laboratorio se muestran en la Tabla 1.
Se suspendió ceftazidima y se comenzó con cefepime.
En el noveno día de hospital, los vómitos aumentaron en frecuencia sin
anorexia ni diarrea. La presión sistólica era de 70 a 80 mm Hg. Los ruidos
intestinales estaban disminuidos de intensidad, y el abdomen estaba levemente distendido
con dolor difuso y signo de rebote.
Se discontinuó tobramicina y se comenzó con metronidazol y líquidos
intravenosos, suspendiéndose la ingesta de sólidos y líquidos por vía oral. Los
resultados de laboratorio se muestran en la tabla 1. Estaban pendientes los
resultados de otros tests. Una radiografía de tórax no se había modificado, y
las radiografías de abdomen revelaban asas no dilatadas de intestino delgado y
grueso sin neumoperitoneo ni obstrucción intestinal.
Una TC de tórax mostró cambios bronquiectásicos leves en ambas bases
pulmonares y un patrón en “árbol brotado” (tree-in-bud opacities ) en la base
pulmonar derecha que se consideró representativo de bronquiolitis. La TC de
abdomen después de la administración de contraste oral reveló marcado
engrosamiento de la pared de la totalidad del colon que se extendía al
intestino delgado que fueron sugestivos de contenido fecal. Infiltración
prominente de la grasa mesentérica y moderada ascitis sin neumatosis o aire
libre. El hígado tenía un contorno nodular sugestivo de cirrosis, y había
reemplazo graso del páncreas; los uréteres y la vejiga eran normales. Durante
las siguientes 12 horas después de a TC el dolor abdominal aumentó y el abdomen
se distendió masivamente. En el décimo dé de hospital se llevó a cabo un
procedimiento diagnóstico, y se recibieron los resultados de algunos
estudios pendientes.
Diagnóstico Diferencial.
Imágenes.
Una radiografía de frente y perfil de tórax del ingreso mostró
hiperexpansión pulmonar con bronquiectasias difusas y distorsión de la
arquitectura bilateralmente, hallazgos todos ya vistos en radiografías
anteriores y compatibles con su diagnóstico de fibrosis quística (Figura 1 A).
No hay opacidades del espacio aéreo sugestivos de neumonía ni derrame pleural.

Figura 1. Radiografías de tórax y abdomen.
A. Radiografía de tórax frente del día de la internación que muestra
pulmones hiperexpandidos (Panel A). Hay bronquiectasias difusas y distorsión
arquitectural bilateral (flechas). La punta del catéter percutáneo se proyecta
en la escápula derecha (cabeza de flecha). Una radiografía de abdomen obtenida
8 días después de la internación (Panel B) muestra leve engrosamiento mucoso
que compromete asas del centro del abdomen (flechas). No hay dilatación
sugestiva de obstrucción. No hay neumoperitoneo.
Una radiografía de abdomen obtenida 8 días después de la internación con el
paciente en decúbito ventral y de pie sugestivos de edema mucoso comprometiendo
asas de la región centroabdominal (Figura 1B). No había evidencias de
neumoperitoneo. No había dilatación de asas sugestivas de obstrucción. La TC de
abdomen y pelvis después de la administración de contraste oral reveló marcado
engrosamiento circunferencial de la pared intestinal desde el recto hasta el ciego (Figura 2A),
con infiltración mesentérica extensa (Figura 2 B), y moderada ascitis (Figura
2C). El engrosamiento mucoso producía haustración de pliegues alternando con
puentes mucosos transversos llamados el “signo del acordeón” ya que recuerda el
instrumento musical (1) (Figura 2C).
Estos hallazgos son consistentes con colitis que afecta la totalidad del
colon. El diagnóstico diferencial incluye la colitis pseudomembranosa, colitis
ulcerosa y colitis isquémica. La apariencia de materia fecal heterogénea dentro
del íleon terminal sugiere la presencia de estasis intestinal, probablemente
relacionado con la fibrosis quística. No hay evidencias de obstrucción. El
páncreas en su totalidad está reemplazado por grasa, hallazgo compatible con el
diagnóstico de fibrosis quística. El hígado tenía un contorno nodular
consistente con cirrosis o fibrosis.

Figura 2 . TC de abdomen obtenida después de la administración de contraste
oral el día 8 de intenación.
Panel A que muestra engrosamiento de la pared colónica desde el recto hasta
el ciego (flechas) y también comprometiendo el intestino delgado. Materia fecal
heterogénea dentro de la luz del ileon terminal (cabezas de flechas)
consistente con el signo de “heces en delgado”, hallazgo sugestivo de estasis
intestinal. Una imagen axial a nivel del ileon terminal (Panel B) muestra
engrosamiento difuso circunferencial de la pared (flecha), y el signo de heces
en el delgado (cabeza de flecha). Una imagen axial a nivel del colon transverso
muestra engrosamiento circunferencial de la pared (Panel C, flechas). El
engrosamiento mucoso es consistente con el “signo del acordeón” (cabeza de
flecha).
¿Cuál es el diagnóstico?
La resolución del caso se publicará el domingo 28/10/2012
Eventos vasculares.
Los eventos vasculares agudos tales como la trombosis oclusivas o no oclusivas raramente evolucionan al compromiso de la totalidad del colon dado la abundante red vascular de este órgano. La trombosis de la porta o las trombosis de las venas mesentéricas son improbables en este caso, y de estar presentes mostrarían también compromiso del intestino delgado. Aunque la TC fue obtenida sin contraste, no hay densidades compatibles con coágulos que se vean en la vasculatura. Sin embargo, en la colitis isquémica raramente se ven coágulos arteriales en la angiografía.
Diagnóstico clínico presuntivo:

Figura 3. Muestra de intestine obtenido en el momento de la cirugía.


Este paciente de 34 años con fibrosis quística se presentó con exacerbación
de su enfermedad pulmonar por lo que fue tratado con múltiples antibióticos. En
el noveno día de internación, el paciente presentó vómitos y se volvió
hipotenso con dolor abdominal difuso. El recuento de blancos estaba elevado. El
hematocrito y el nivel de creatinina aumentaron, datos en favor de depleción de
volumen intravascular. Desarrolló acidosis láctica. Se agregó metronidazol al
tratamiento. La TC mostró hallazgos compatibles con pancolitis; dentro de las
12 horas el abdomen evolucionó a distensión masiva con aumento del dolor.
Hay dos preguntas que hacerse en este punto. Primera, ¿qué está causando
este cuadro agudo del paciente? dado su rápido deterioro, y la segunda, ¿hay
pérdida de la viabilidad intestinal que requiere cirugía de emergencia?
Nos referiremos primero a la enfermedad aguda del paciente y su diagnóstico
diferencial y posteriormente consideraremos los datos de la historia y del
examen que son indicaciones de exploración quirúrgica inmediata.
El diagnóstico diferencial del cuadro agudo de este paciente puede ser
agrupado en cuatro áreas o formas de compromiso: compromiso vascular,
obstrucción mecánica, inflamación e infección.
Los eventos vasculares agudos tales como la trombosis oclusivas o no oclusivas raramente evolucionan al compromiso de la totalidad del colon dado la abundante red vascular de este órgano. La trombosis de la porta o las trombosis de las venas mesentéricas son improbables en este caso, y de estar presentes mostrarían también compromiso del intestino delgado. Aunque la TC fue obtenida sin contraste, no hay densidades compatibles con coágulos que se vean en la vasculatura. Sin embargo, en la colitis isquémica raramente se ven coágulos arteriales en la angiografía.
Obstrucción.
Este paciente tenía antecedentes de síndrome obstructivo del intestino
delgado distal, complicación que ocurre en el 10 a 20% de los pacientes con
fibrosis quística. La fisiopatología es la misma que la del íleo meconial
(falta de pasaje de meconio desde el colon dentro de las 48 horas del
nacimiento), que es la primera manifestación de la fibrosis quística y que se
ve en el 15% de los recién nacidos con
esta enfermedad. La mutación en el gen CFTR (que codifica para el canal
epitelial de cloro en la membrana apical plasmática) (2), lo que lleva a la
formación de secreciones espesas en órganos que presentan secreción luminal
(pulmones, intestino, ductos pancreáticos, y tracto reproductor masculino), y a
una excesiva respuesta inflamatoria del huésped lo que resulta en el caso del
intestino, un dificultoso pasaje de materia fecal. Los pacientes con síndrome
de obstrucción distal del intestino delgado tienen crisis recurrentes de dolor
abdominal (típicamente en el cuadrante inferior derecho del abdomen), y
constipación.(3) El síndrome puede también afectar el colon o el recto lo que
resulta en constipación crónica y megacolon adquirido. Aunque este paciente tenía este síndrome en
el pasado, el súbito comienzo de engrosamiento de la pared del colon, y la
forma tóxica de presentación no es compatible con este diagnóstico. No es raro
que los pacientes con fibrosis quística tengan pobres movimientos gástricos e
intestinales lo que lleva a sobrecrecimiento bacteriano, lo que a su vez puede
causar dolor y distensión. Sin embargo, este cuadro también se presenta de una
manera más crónica, en contraste con la presentación aguda de la enfermedad
actual de este paciente, que además no explica los hallazgos radiológicos.
El dolor abdominal intermitente en pacientes con fibrosis quística puede
ser resultado de la intususcepción en la región ileocecal, en la cual la materia
fecal espesada actúa como cabeza de invaginación. (4) Sin embargo, este
paciente no tuvo el clásico signo de doughnut (rosquilla) o del target (blanco
de tiro) del intestino edematoso contrastando con la grasa en la TC.
El prolapso rectal se ve en el 20% de los pacientes con fibrosis quística,
como resultado de la voluminosa materia fecal y de la hipotonía muscular. Sin
embargo, esta condición es rara en adultos y no fue vista en el examen de este
paciente. También el riesgo de cáncer en pacientes con fibrosis quística es
siete veces mayor que una persona sin fibrosis quística. El cáncer se encuentra
más comúnmente en el tubo digestivo (5), e incluye adenocarcinomas de esófago,
intestino delgado y vía biliar y sobre todo adenocarcinomas de colon y recto
que pueden verse tan tempranamente como a los 13 años de edad. La TC no mostró
evidencias de tumor o de una obstrucción en un punto en este caso.
Inflamación.
En adultos asintomáticos con fibrosis quística, los estudios de imágenes
del colon frecuentemente muestran grados variables de engrosamiento de la
pared. La pneumatosis intestinal, que se encuentra confinada al colon se ve en
aproximadamente en 5% de los pacientes y está frecuentemente asociada a
enfermedad pulmonar obstructiva crónica. Así, los hallazgos anormales del colon
en la TC no deben ser indicación de evento agudo y hay que establecer una buena
correlación clínica para darle una interpretación a esos hallazgos
radiológicos en un adecuado contexto. En
este caso, las imágenes se correlacionan con el empeoramiento de la condición
clínica del paciente y son sugestivos de colitis severa.
La apendicitis es infrecuente en pacientes con fibrosis quística a pesar
del hecho de que el apéndice está crónicamente distendido en estos pacientes.
La prevalencia de la enfermedad de Crohn pero no de colitis ulcerosa puede ser
mayor en estos pacientes que en la población general. (6)
La colopatía fibrosante, una complicación de la suplementación con altas
dosis de enzimas pancreáticas puede presentarse con engrosamientos o
estrecheces típicamente en colon derecho debido a fibrosis submucosa. (7,8) Los
pacientes pueden referir síntomas obstructivos, diarrea sanguinolenta, o ambas
cosas. Los estudios de imágenes muestran engrosamiento de la pared del colon con
estrechamiento luminal con compromiso más notable del colon ascendente. Con
excepción de la colitis ulcerosa que puede ser causa de megacolon tóxico,
ninguno de estos diagnósticos es probable en este caso.
Infección.
Este paciente puede tener colitis infecciosa? La colitis por
citomegalovirus (CMV) es improbable dado que este paciente no está
inmunocomprometido y no ha tenido antecedentes de infección primaria por CMV
que generalmente se expresa como síndrome mononucleósico con lesiones cutáneas,
aumento de transaminasas y linfocitosis atípica.
Las infecciones bacterianas del intestino deben ser consideradas incluyendo
salmonella, shigella, y campylobacter, pero todas ellas raramente pueden
producir megacolon tóxico. Sin embargo, este paciente ha sido tratado con
múltiples antibióticos en un ámbito hospitalario. La combinación de esos dos
factores, aumenta el riesgo de desarrollo de colitis por Clostridium.
Difficile. La colitis por Clostridium difficile es rara en pacientes con
fibrosis quística, aunque el estado de portador asintomático ha sido reportado
en 22 a 50% de tales pacientes. (9,10) La rareza de la colitis declarada puede ser explicada en estos pacientes por la
resistencia de las células epiteliales colónicas a distinto tipo de toxinas como por ejemplo
la del cólera. (119 Aunque la literatura muestra sólo pocos casos de colitis
por C. difficile en pacientes con fibrosis quística (12,13,14), en cada uno de estos casos publicados resalta
el curso fulminante, con distensión y dolor abdominal progresivos, y
ausencia de diarrea. La febrícula y la leucocitosis son comunes. La TC revela pancolitis e infiltración
paracolónica, y en un caso reportado se menciona ascitis y el “signo del
acordeón”. (15) Aunque el “signo del acordeón” puede ser visto en cualquier
forma de colitis severa, en una serie de cinco pacientes con fibrosis quística
y colitis por C. diffile, tres tenían atrapamiento de gas dentro de los
pliegues lo que producía el signo del acordeón; en este paciente dicho signo
fue observado.
La tasa de mortalidad reportada es
de aproximadamente 50%. La mayoría de los pacientes en quienes la laparotomía
exploradora con colectomía subtotal pudo ser realizada sobrevivieron.
La infección por C. difficile con colitis fulminante y megacolon tóxico es
altamente probable en este paciente. El uso de antibióticos de amplio espectro
incluyendo clindamicina, fluoroquinolonas, y cefalosporinas se asocian al
desarrollo de colitis por C. difficile. Similar a los casos reportados de
fibrosis quística en la literatura, este paciente se presentó con colitis
fulminante, sin diarrea, y rápida progresión a megacolon tóxico. El diagnóstico
puede ser hecho por el envío de una muestra de materia fecal para análisis de
la toxina. Aunque la mayoría de los tests que detectan toxina de C. difficile
tienen una especificidad de 99%, tienen una sensibilidad de 60%. (16) El examen
endoscópico con el hallazgo de pseudomembranas puede ser usado como ayuda en el
diagnóstico cuando el paciente tiene severa enfermedad y la muestra de materia
fecal es negativa o hay una mala
respuesta al tratamiento. El ocasional respeto por el rectosigma hace necesaria
una colonoscopía más que una sigmoidoscopía. Sin embargo, en pacientes con
severa colitis, la introducción de aire en el intestino puede causar
perforación y por eso está contraindicada.
Manejo de la colitis por C. difficile.
En este caso, varios hallazgos sugieren isquemia colónica severa o necrosis
y deben hacer que se considere una cirugía de emergencia. Ningún signo de
irritación peritoneal en el examen físico fue mencionado, pero había distensión
masiva del abdomen lo cual debe ser causa de preocupación de la presencia de
necrosis. Otro indicador de pérdida de viabilidad es la evidencia de
extravasación de líquidos lo que indicaría isquemia o necrosis. Cuando esto
ocurre, una disminución del volumen intravascular se acompaña de un aumento en
los niveles de hematocrito, y si es lo suficientemente severo, también aumentos
de urea y creatinina. El déficit intravascular puede ser groseramente
calculado de la siguiente forma: restar el hematocrito basal del hematocrito
actual y dividir los resultados por el hematocrito basal; a esa cifra se la
multiplica por un tercio del peso corporal en kg.
Este paciente pesaba aproximadamente 55 kg; su hematocrito en la admisión
al hospital era de 40 y aumentó a 52, lo que refleja un déficit de volumen de
5,5 litros. Esto explica el desarrollo de hipotensión arterial y fallo renal, y
es indicación de medidas de resucitación con expansión rápida. La adecuada
repleción de volumen puede ayudar a salvar regiones isquémicas del intestino de
la necrosis por hipoperfusión.
Resumen.
En este paciente se produjo una pancolitis en el contexto de la
administración de múltiples antibióticos. Su curso clínico y estudios de
imágenes son consistentes con el diagnóstico de infección fulminante por C.
difficile. El rápido deterioro del estado general, incluyendo el shock,
distensión abdominal masiva, y valores de laboratorio consistentes con severa
pérdida de volumen intravascular, sugieren pérdidad de viabilidad colónica.
Aunque el análisis de la materia fecal para toxina de C. difficile puede ser de
ayuda, ni un resultado positivo, ni un resultado negativo serían diagnósticos
dado la alta tasa de portadores entre pacientes con fibrosis quística, y la
baja sensibilidad de la prueba. La sigmoidoscopía con sigmoidoscopio flexible
mostraría pseudomembranas que confirmarían el diagnóstico. Sin embargo, en
vista del rápido deterioro del estado general en el contexto de megacolon
tóxico, una colectomía subtotal de emergencia se impone para salvar la vida del
paciente. Aunque un apropiado esquema terapéutico con antibióticos es curativo
en la mayoría de los casos de infección, un día de terapia con metronidazol intravenoso
ya había sido administrado en este
paciente, y el daño colónico era demasiado avanzado para responder a los
antibióticos.
Colitis fulminante por C. difficile con megacolon tóxico.
Discusión patológica.
Una muestra de material fecal obtenida el 9° día de internación fue
positivo para toxina de C. difficile. Al día siguiente se llevó a cabo una
ileocolectomía. La pieza consistía en un segmento de colon de 171 cm, y un
segmento de 5 cm de íleon terminal. El colon estaba distendido y la pared
estaba engrosada (Figura 3A). La superficie mucosa a todo lo largo del colon y
del íleon terminal estaba cubierta por un material amarillo-verdodo que se
despegaba fácilmente, hallazgo consistente con pseudomembranas. (Figura 3A
inserto). 
Una fotografía de la macroscopía del colon resecado muestra dilatación
masiva (Panel A); la superficie mucosa de la totalidad del colon y del íleon
terminal estaba cubierto por una capa de material fibrinoide amarillo-verdoso
que era fácilmente despegado, consistente con pseudomembranas (inserto). Una
vista a bajo aumento de la pared colónica (Panel B hematoxilina-eosina),
muestra marcado engrosamiento causado principalmente por edema submucoso con
congestión; la muscularis propia está atenuada debido a necrosis. La capa
mucosa muestra lámina propia edematosa con infiltración celular, así como
criptas distendidas por mucina y células inflamatorias cubiertas por
pseudomembranas (Panel C, hematoxilina-eosina). A gran aumento, se muestra la
descamación de las células epiteliales y estructuras “en fantasma”
emtremezcladas con fibrina y neutrófilos (Panel D, hematoxilina-eosina).
En el examen microscópico, el engrosamiento de la pared colónica era
especialmente debido a edema y congestión de la submucosa (Figura 3B). Había un
exudado fibrinopurulento en la superficie mucosa que en áreas puede ser visto
extruyendo las criptas mucosas con un aspecto volcánico consistentes con
pseudomembranas vistas macroscópicamente. La superficie de las criptas estaba
distendidas por mucina y células inflamatorias (Figura 3C). Había necrosis y
células epiteliales descamadas entremezcladas con fibrina y neutrófilos (Figura
3D). El diagnóstico anatomopatológico es de colitis pseudomembranosa y
enteritis, lo cual es consistente con enterocolitis por C. difficile.
En las primeras 24 horas de post operatorio la condición del paciente
mejoró, con un descenso en el recuento de glóbulos blancos, y corrección de la
acidosis. Sin embargo, en el 11° día de internación, después de la extubación
de la tráquea, se produjo un episodio de disnea, hipotensión, y acidosis, que
requirió reintubación y soporte con vasopresores. En las siguientes 24 horas
presentó hipotensión refractaria y acidosis láctica, el recuento de glóbulos
blancos aumentó a 90.200/mm3 con 28% de formas en banda, aumento de las
aminotransferasas, coagulación intravascular diseminada y fallo renal agudo. El
día 12° sufrió un paro cardiorrespiratorio y las maniobras de resucitación no
tuvieron éxito. Se llevó a cabo una autopsia.
En la autopsia se vio cómo quedaba un corto segmento de colon que había
sido anastomosado al íleon. El íleon estaba distendido con adherencias serosas.
Tanto el colon como la mayor parte de la mucosa ileal tenían pseudomembranas, y
el examen microscópico mostró similar morfología que la vista en la pieza
quirúrgica de ileocolectomía, con degeneración más avanzada de las criptas del
epitelio.
Los pulmones mostraban cambios consistentes en fibrosis quística avanzada
(Figura 4A), incluyendo lesiones quísticas representando neumatoceles derivados
de viejos abscesos y bronquiectasias y bronquiolectasias con fibrosis
peribronquial y peribronquiolar desde los grandes bronquios a los bronquiolos
respiratorios, con moco viscoso en la luz dilatada (Figura 4B). La fibrosis se
extendía al parénquima pulmonar peribronquial y peribronquiolar, con áreas de
consolidación pero sin áreas de bronconeumonía activa.

Figura 4. Hallazgos en la autopsia.
Una fotografía de la macroscopía de
los pulmones muestra numerosos espacios aéreos dilatados con paredes
engrosadas y varias lesiones quísticas grandes como resultado de viejos
abscesos de pulmón (Panel A). Áreas de consolidación mal definidas son también
evidentes en lóbulos inferiores. Los espacios aéreos desde los bronquiolos
terminales. Hasta los bronquiolos respiratorios están extensamente dilatados
(bronquiolectasas), con moco viscoso en la luz (Panel B); también se ve
fibrosis peribronquiolar. El páncreas está casi totalmente reemplazado por
grasa (Panel C). Hay puentes fibróticos en el hígado (Panel D, hematoxilina
eosina), con proliferación de conductos biliares que contienen gruesas
secreciones características de la fibrosis quística (inserto).
El daño hepático en la fibrosis quística es debida a la bilis anormalmente
espesa que se acumula en el tracto biliar causando inflamación y subsecuente
pérdida de parénquima. En este caso había puentes fibróticos con esteatosis
leve (Figura 4 C), pérdida de los conductos biliares en algunos espacios
portales, y proliferación de dúctulos biliares que contenían secreciones
eosinofílicas amorfas (Figura 4 C inserto), todas las cuales son
características de la fibrosis quística. El páncreas estaba atrófico
reemplazado por tejido fibroso y graso (Figura 4D), como resultado de
secreciones espesas en el sistema ductal pancreático que causan inflamación y
atrofia parenquimatosa.
La causa de la muerte en este caso fue enterocolitis pseudomembranosa con
septicemia en un paciente con fibrosis quística.
Diagnóstico anatomopatológico:
Colitis pseudomembranosa y enteritis, hallazgos consistentes con
enterocolitis por C. difficile.
Fibrosis quística asociada con bronquiectasias pulmonares, fibrosis
hepática y atrofia pancreática.
Conclusiones del caso.
“Ay, de aquel niño que al ser besado en la frente sabe salado. Él está
embrujado y pronto debe morir”. Esta frase del folklore irlandés del siglo XV,
atribuía el sabor salado del sudor a una brujería. Era común en el pasado
atribuir el exceso de sal en la frente de los niños a hechizos, encantos, magia, o posesión demoníaca. "...Una señora honorable dice que conoce
a la gente embrujada, si al rascarles la frente, uno nota después un sabor
salado en los dedos...".
En el año 1905 Karl Landsteiner describió la asociación entre el meconio
espeso en un recién nacido con fibrosis pancreática a raíz de un caso de íleo
meconial. En 1912 Sir Archibald Garrod describió familias, algunos de cuyos
niños tenían esteatorrea y se morían de bronconeumonía sugiriendo un modelo
recesivo de herencia. En 1936, Guido Fanconi, un pediatra suizo, fue el primero
en usar el término fibrosis quística (FQ) para describir la combinación de
insuficiencia pancreática y enfermedad pulmonar crónica en niños. Carl von
Rokitansky describió un caso de muerte fetal con peritonitis meconial, una
complicación del íleo meconial asociado con la fibrosis quística. Pero fue
recién en 1938 en que Dorothy Andersen
publicó un artículo intitulado “Cystic fibrosis of the páncreas and its
relation to celiac disease: a clinical and pathological study”("La
fibrosis quística del páncreas y su relación con la enfermedad celíaca: un
estudio clínico y patológico") en la revista American Journal of Diseases
of Children. De esta manera, era la primera investigadora en definir esta
entidad nosológica (denominada, por aquel entonces, "fibrosis quística del
páncreas"), y en correlacionarla con los trastornos pulmonares e
intestinales prominentes. También postuló que era una enfermedad recesiva y
utilizó el reemplazo de enzimas pancreáticas como tratamiento para los niños
afectados.

Figura. Dorothy H. Andersen, quien
describió por primera vez la fibrosis quística
(National Library of Medicine)
En 1952, Paul di Sant' Agnese
descubrió anomalías en los electrolitos del sudor. Sobre la base de esa
evidencia, se desarrolló y perfeccionó el examen del sudor durante el curso de
la siguiente década. En 1943, Sydney Farber reconoce a la FQ como una
enfermedad sistémica, y acuña el término “mucoviscidosis” (del lat. muccus,
"moco", y viscōsus, "pegajoso").
Todavía en los años 60, la perspectiva para los niños afectados continuaba
siendo terrible pues la mayoría moría durante la niñez, después de años de
sufrimiento. Recién en 1976, Mitchell-Heggs reporta los primeros 45 pacientes
mayores de 12 años, procedentes de 3 hospitales de Londres. Sólo en 1979 se
pudo disponer de screening neonatal mediante la utilización de tripsina
immunorreactiva (IRT). En 1983 Paul Quinton, portador de FQ, publica que la
impermeabilidad al cloro que él había demostrado en las glándulas de sudor, era
la base para la elevación de los electrolitos en el sudor de los pacientes con
FQ. Esto se consideró un paso decisivo en la comprensión del defecto básico. En
1989, el grupo de Lap-Chee Tsui identifica y clona el gen de la FQ, ubicado en
el brazo largo del cromosoma 7, y a la proteína para la cual codifica, la
denomina proteína de conductancia transmembrana de la FQ (CFTR, por su sigla en
inglés), publicando los hallazgos en la revista Science. La demostración que el tratamiento precoz y
agresivo mejora el pronóstico obliga a mejorar la precocidad del diagnóstico
(tamizaje neonatal) y hacer los mayores esfuerzos en el desarrollo de avances,
tanto en las terapias convencionales (farmacológico, nutricional, etc.), como
en las más innovadoras (trasplante pulmonar, terapia génica).
Este paciente de 34 años analizado aquí, estaba a la espera de un
transplante pulmonar, cuando se injertó una complicación que finalmente lo
llevó a la muerte después de un penoso curso de su enfermedad por más de tres
décadas, y que había debutado a las 48 horas de su nacimiento con un íleo
meconial (complicación observada en 10 a 20% de los pacientes con FQ), que requirió cirugía. Desde entonces,
sucesivas infecciones especialmente del aparato respiratorio habían sido una
constante de su evolución, y la imposibilidad de erradicación de gérmenes de
extrema virulencia y resistencia creciente como Pseudomona aeruginosa y
Staphylococcus aureus meticilino resistente, habían sido responsables de la
declinación de su función respiratoria y de la necesidad de utilización
obligada de múltiples esquemas de antibióticos para su control. Justamente la
utilización crónica de antibióticos de amplio espectro en pacientes con FQ es
la causa de la alta tasa de colonización por Clostridium difficile en ellos,
que va de 20 a 50%. Paradójicamente es raro que pacientes con FQ presenten
infección clínica por C. difficile, fenómeno que tendría explicación en cierta
resistencia de las células epiteliales del colon a algunas toxinas como la de
este gérmen, así como a otras toxinas
como la colérica. Esta resistencia no fue suficiente sin embargo, para que este
paciente presentara una infección fulminante por Clostridium difficile
seguramente como resultado de la múltiple asociación de antibióticos en el
contexto de internación de más de una semana, que determinaran una cirugía
resectiva (ileocolectomía), seguida de sepsis fatal.
Es difícil tener una clara comprensión de cómo, una alteración de la
composición y de la densidad del moco en algunos órganos como pulmón, páncreas,
intestino e hígado, pueda conducir a tamaño deterioro orgánico general con
impresionante impacto en la calidad y expectativas de vida. Esta alteración de la calidad y composición
del moco asienta en la mutación del gen CFTR (Cystic fibrosis transmembrane
conductance regulator), que codifica para una proteína, la proteína CFTR.

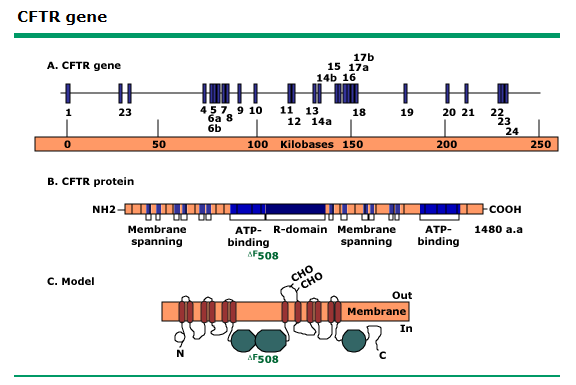
Figura. Gen CFTR. Representación
esquemática del gen CFTR y su polipéptido codificado. ΔF508 se refiere al sitio
más común de la mutación que causa fibrosis quística
La proteína, sintetizada a partir del gen CFTR es una proteína de 1480
aminoácidos que transporta iones cloruro a través de las células epiteliales,
se une a la membrana externa de las células de las glándulas sudoríparas,
pulmón, páncreas, y otros órganos afectados, atraviesa esta membrana y actúa
como un canal iónico conectando la parte interna de la célula (citoplasma) con
el fluido extracelular. Este canal es
responsable de controlar el paso de cloruro hacia (y desde) el medio
interno. Cuando la proteína CFTR no funciona correctamente, este movimiento se
ve restringido, reteniéndose cloruro en el espacio extracelular. Debido a que
el cloruro tiene carga eléctrica negativa, los iones con carga positiva tampoco
pueden cruzar la membrana citoplasmática, a causa de la atracción
eléctrostática ejercida por los iones cloruro. El sodio es el más abundante de
los iones del medio extracelular, y la combinación de sodio y cloruro da lugar
al cloruro de sodio, el cual se pierde en grandes cantidades en el sudor de los
individuos con FQ. La mayoría de las personas
sanas tienen dos copias funcionales del gen CFTR, y sólo una es
necesaria para impedir el desarrollo de fibrosis quística. La FQ se desarrolla
por lo tanto, cuando ninguno de estos
genes opera normalmente. En consecuencia, se la considera una enfermedad
autosómica recesiva.
A la mayoría de los niños se les diagnostica fibrosis quística antes del
primer año de vida, cuando la mucosidad pegajosa que afecta pulmones y
páncreas, comienza a mostrar su impacto. En el tracto respiratorio, esas
secreciones sirven como caldo de cultivo para diversas bacterias responsables
de infecciones crónicas, con deterioro progresivo y permanente del parénquima
pulmonar. Conforme se agrava la condición respiratoria, los pacientes sufren
hipertensión pulmonar. Por otra parte, en el páncreas, el moco obstruye el
tránsito de las enzimas sintetizadas por la glándula e impide que lleguen hasta
los intestinos con la consiguiente malabsorción.
La enfermedad pulmonar resulta del bloqueo de las vías aéreas más pequeñas
por moco espeso. La inflamación y la
infección producen daño a los pulmones y cambios estructurales que conducen a
una variedad de síntomas. En las etapas iniciales, comúnmente se presentan tos
persistente, producción copiosa de secreciones, y una disminución en la
capacidad aeróbica. Muchos de estos síntomas ocurren cuando ciertas bacterias
(fundamentalmente, Pseudomonas aeruginosa)
que normalmente viven en el moco espeso, crecen en forma descontrolada y causan
neumonías a repetición. A ciencia cierta no se conoce la patogénesis de la
predilección de la infección de las secreciones respiratorias por especies de
Pseudomonas En estados avanzados de la FQ, los cambios en la arquitectura del
pulmón producen dificultades respiratorias crónicas.
Otros síntomas incluyen expectoración de sangre o esputo sanguinolento,
dilatación crónica de los bronquios o bronquiolos (bronquiectasia), elevación
de la presión sanguínea en el pulmón, insuficiencia cardíaca, insuficiencia
respiratoria y atelectasias. Además de
las infecciones bacterianas más comunes, las personas con FQ desarrollan con
mayor facilidad otros tipos de enfermedades respiratorias. Entre éstas se
encuentra la aspergilosis broncopulmonar alérgica, caracterizada por una
respuesta de hipersensibilidad
hongos del género Aspergillus (Aspergillus fumigatus), que agudiza los
problemas respiratorios. Otro ejemplo es la infección por el complejo Mycobacterium avium (MAC), grupo de
actinobacterias emparentadas con Mycobacterium
tuberculosis, que puede ocasionar daños mayores al pulmón, y que no
responde a la terapéutica con antibióticos convencionales.
El karma de gran parte de la vida de este paciente fue Pseudomona aeruginosa,
germen que por distintas razones se adapta muy bien al espeso moco de
las secreciones respiratorias de pacientes con FQ, y de la que no pudo
desembarazarse nunca. Una reciente y atractiva teoría argumenta que una de las
causas de la predisposición a la colonización crónica por P. aeruginosa es la avidez por el oxígeno de las células
epiteliales respiratorias, lo que da por resultado una tensión de oxígeno
disminuido dentro de la capa mucosa hiperviscosa. Esta hipoxia local es un
excelente caldo de cultivo para ese y otros gérmenes. Todo esto motivó el uso
sucesivo y crónico de diferentes antibióticos por distintas vías.
Fluoroquinolonas, cefalosporinas, clindamicina, tobramicina, y muchos otros,
todos descriptos como causantes de infección por C. difficile quien a través de su complicación más temida generó la
puerta de entrada para la sepsis terminal.
CASE RECORDS OF THE MASSACHUSETTS GENERAL HOSPITAL
Fuente
From the Department of Medicine, Beth Israel Deaconess
Medical Center (S.D.F.); the Departments of Radiology (R.N.U.) and Pathology
(M.M.-K.), Massachusetts General Hospital; and the Departments of Medicine
(S.D.F.), Radiology (R.N.U.), and Pathology (M.M.-K.), Harvard Medical School —
all in Boston.
References
1. Macari M, Balthazar EJ,
Megibow AJ. The accordion sign at CT: a
nonspecific finding in patients with colonic edema. Radiology 1999;211:743-746.
[Free Full Text]
2. Accurso
FJ. Update in cystic fibrosis 2007. Am J Respir Crit Care Med
2008;177:1058-1061. [Free Full Text]
3. Dray
X, Bienvenu T, Desmazes-Dufeu N, Dusser D, Marteau P, Hubert D. Distal
intestinal obstruction syndrome in adults with cystic fibrosis. Clin
Gastroenterol Hepatol 2004;2:498-503. [CrossRef][Medline]
4. Robertson
MB, Choe KA, Joseph PM. Review of the abdominal manifestations of cystic
fibrosis in the adult patient. Radiographics 2006;26:679-690. [Free Full Text]
5. Maisonneuve
P, FitzSimmons SC, Neglia JP, Campbell PW III, Lowenfels AB. Cancer risk in
nontransplanted and transplanted cystic fibrosis patients: a 10-year study. J
Natl Cancer Inst 2003;95:381-387. [Free Full Text]
6. Lloyd-Still
JD. Crohn's disease and cystic fibrosis. Dig Dis Sci 1994;39:880-885.
[CrossRef][Web of Science][Medline]
7. Schwarzenberg
SJ, Wielinski CL, Shamieh I, et al. Cystic fibrosis-associated colitis and
fibrosing colonopathy. J Pediatr 1995;127:565-570. [CrossRef][Web of
Science][Medline]
8. Smyth
RL, Ashby D, O'Hea U, et al. Fibrosing colonopathy in cystic fibrosis: results
of a case-control study. Lancet 1995;346:1247-1251. [CrossRef][Web of
Science][Medline]
9. Peach
SL, Borriello SP, Gaya H, Barclay FE, Welch AR. Asymptomatic carriage of
Clostridium difficile in patients with cystic fibrosis. J Clin Pathol
1986;39:1013-1018. [Free Full Text]
10. Welkon
CJ, Long SS, Thompson CM Jr, Gilligan PH. Clostridium difficile in patients
with cystic fibrosis. Am J Dis Child 1985;139:805-808. [Free Full Text]
11. Gabriel
SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote
resistance to cholera toxin in the cystic fibrosis mouse model. Science
1994;266:107-109. [Free Full Text]
12. Binkovitz LA, Allen E, Bloom
D, et al. Atypical presentation of
Clostridium difficile colitis in patients with cystic fibrosis. AJR Am J
Roentgenol 1999;172:517-521. [Free Full Text]
13. Rivlin
J, Lerner A, Augarten A, Wilschanski M, Kerem E, Ephros MA. Severe Clostridium
difficile-associated colitis in young patients with cystic fibrosis. J Pediatr
1998;132:177-179. [CrossRef][Web of Science][Medline]
14. Yates
B, Murphy DM, Fisher AJ, et al. Pseudomembranous colitis in four patients with
cystic fibrosis following lung transplantation. Thorax 2007;62:554-556. [Free
Full Text]
15. Kawamoto
S, Horton KM, Fishman EK. Pseudomembranous colitis: spectrum of imaging
findings with clinical and pathologic correlation. Radiographics
1999;19:887-897. [Free Full Text]
16. Blossom
DB, McDonald LC. The challenges posed by reemerging Clostridium difficile
infection. Clin Infect Dis 2007;45:222-227. [CrossRef][Web of Science][Medline]


