En este ejercicio clínico se presenta
un caso que es discutido por un médico internista al que se le van
proporcionando datos de la historia clínica en forma secuencial, y este analiza
el cuadro a la luz de los nuevos elementos, de una manera análoga al proceso
diagnóstico en la práctica real de la medicina.
Un hombre de 60
años se presentó al departamento de emergencias para evaluación de sangrado
rectal, síncope, y dolor en pierna derecha. Cinco días antes había aparecido un
dolor abdominal difuso que aumentaba con los movimientos, asociado a náuseas,
anorexia, y malestar general. El paciente no había viajado recientemente ni
ingerido alimentos inusuales para él, no refería fiebre, pérdida de peso, ni
alteraciones en el tránsito intestinal. Aproximadamente seis horas antes de la
admisión presentó un episodio de hematoquecia y hematuria seguido de síncope.
Inmediatamente después y al recuperarse espontáneamente del síncope presentó
intenso dolor en pierna derecha por lo que buscó ayuda médica.
Este paciente se presentó con elementos de múltiples sistemas por lo que
un diagnóstico unificador no surge claramente en forma inmediata. Un sangrado
digestivo bajo en este grupo etario se puede explicar por diverticulosis,
colitis (isquémica, infecciosa, o
inflamatoria), carcinoma, o malformaciones vasculares. El dolor
abdominal asociado apoya más al diagnóstico de colitis isquémica o enfermedad
inflamatoria intestinal. El dolor no es frecuente en la diverticulosis; es
común en la diverticulitis pero en ese caso no es frecuente el sangrado. La
ausencia de fiebre hace improbable el diagnóstico de infección. La presentación
súbita, y la ausencia de pérdida de peso o cambios en el hábito intestinal
argumentan en contra de enfermedad neoplásica. La aparición de síncope puede
reflejar una reacción vasovagal, pero despierta sospecha de reducción
sustancial en el volumen plasmático.
Las típicas causas de hematuria macroscópica tales como nefrolitiasis,
infección urinaria, o carcinoma de vejiga no son causa de hematoquecia. Una
condición que puede explicar ambos hallazgos, así como el dolor abdominal es
una fístula enterovesical relacionada con enfermedad inflamatoria intestinal o
enfermedad diverticular.
El severo dolor en la pierna es también preocupante. Si el paciente
hubiese tenido una enfermedad vascular periférica previa, un cuadro de
hipovolemia causado por hemorragia podría haber exacerbado su insuficiencia
vascular. Alternativamente el cáncer de colon o de vejiga, pueden asociarse a
estados hipercoagulables que lo predispongan a la trombosis venosa. Puede ser
que todos estos elementos que presenta el paciente no se expliquen por un único
diagnóstico. Una evaluación de signos los vitales, un completo examen físico y
un laboratorio son urgentemente necesarios.
El paciente tenía
antecedentes de diarrea crónica sin sangre de dos años de evolución. Un año
antes de su presentación actual él se había sometido a una colonoscopía que
había mostrado diverticulosis y dos pólipos. Los pólipos fueron extraídos y
demostraron ser benignos. El resultado de una seriada gastrointestinal alta seguido de una
endoscopía digestiva alta fueron normales. Se comenzó un tratamiento empírico
con metronidazol por posible giardiasis asociado a hioscina por posible colon
irritable los cuales no mejoraron el cuadro.
El paciente también
tenía antecedentes de hipertensión arterial, hiperlipemia, y gota. Su
medicación incluía atenolol 50 mg/día, atorvastatina 20 mg/día, allopurinol 300
mg/día, y aspirina 81 mg /día. Su padre había fallecido de cáncer de colon a
edad avanzada. El paciente tenía antecedentes remotos de abuso de alcohol pero
no tomaba desde hacía 20 años. Había sido un fumador moderado pero
recientemente había dejado de hacerlo.
No queda claro si existe alguna relación entre la diarrea crónica del paciente y su
enfermedad actual. La enfermedad inflamatoria intestinal sigue siendo una
posible asociación entre las dos pero el dato de una colonoscopía negativa está
muy en contra de esta hipótesis diagnóstica. El antecedente de hipertensión,
hiperlipemia y tabaquismo lo predisponen a la enfermedad vascular y por eso
debe considerarse a la insuficiencia vascular como causa del dolor en la
pierna. Un nivel disminuido de hemoglobina asociado a hemorragia puede
contribuir al riesgo de insuficiencia
vascular. La terapia con aspirina lo predispone al riesgo de sangrado digestivo
alto por hemorragia gastrointestinal que si es importante y rápido puede manifestarse
por hematoquecia, explicando así el cuadro de este paciente.
El estudio de una diarrea crónica parece haber sido insuficiente y
debiera haber sido más agresivo. En relación a la enfermedad actual de este
paciente, su antecedente de diverticulosis colónica aumenta la probabilidad de una
diverticulitis. El paciente tenía una historia familiar de cáncer de colon; sin
embargo, los resultados negativos de la colonoscopía hacen que este diagnóstico
sea improbable. Los factores de riesgo vascular de este paciente deben hacernos
considerar a la isquemia intestinal como probable origen del dolor abdominal y
la sangre en la materia fecal.
En el examen
físico, el paciente estaba alerta pero con compromiso general moderado. Era
obeso; su peso 108,4 kg, y su altura era de 175 cm con un BMI de 35,4. Estaba
afebril; la presión arterial era de 74/40 mm Hg y la frecuencia cardíaca de 74
por minuto. La saturación de oxígeno era de 98% mientras respiraba aire
ambiente.
Estaba desdentado,
tenía prótesis dentaria y las mucosas secas. El examen cardíaco reveló una
frecuencia y ritmos regulares sin soplos ni galope. No había ingurgitación
yugular ni soplos en cuello. El abdomen era difusamente doloroso pero no había
reacción peritoneal, masas pulsátiles ni dolor a la descompresión. Los ruidos
intestinales eran normales. El examen rectal mostró un esfínter tónico y sangre
roja pero no era doloroso ni se palpaban masas. Las piernas no dolían, ambas
impresionaban calientes pero la derecha tenía pulsos disminuidos respecto de la
izquierda. El examen neurológico no reveló déficits focales.
La hipotensión puede ser explicada por hipovolemia secundaria a
hemorragia, o a sepsis relacionada con infección intraabdominal. Si hay una
infección, una coagulopatía asociada puede haber potenciado o exacerbado el
sangrado digestivo. La inadecuada frecuencia cardíaca compensatoria puede ser
explicada por el uso de betabloqueantes. Los hallazgos semiológicos en la
pierna derecha sugieren la presencia de flujo vascular disminuido que podría
ser explicado por el bajo gasto cardíaco asociado a enfermedad vascular
periférica. Es necesaria una inmediata resucitación con líquidos con monitoreo
en una unidad de cuidados intensivos. Una evaluación inmediata del origen de la
hemorragia digestiva y mejoramiento de la perfusión de la pierna derecha son
necesarios.
La presencia de ruidos intestinales y ausencia de dolor a la
descompresión o defensa, descartan peritonitis y perforación libre. Una
perforación confinada o bloqueada sin
embargo no puede ser descartada con estos elementos. La disminución de pulsos
en la pierna derecha combinado con hematoquecia aumentan la posibilidad de
fístula arterientérica. Tales fístulas se manifiestan típicamente por
hemorragia gastrointestinal intermitente, signos de sepsis o ambos. Los
factores de riesgo para este tipo de fistulización incluyen injerto vascular
protésico, aneurismas, úlceras penetrantes, tumores, radioterapia, trauma,
diverticulitis e ingestión de cuerpo extraño. El paciente no tenía ninguno de
estos factores de riesgo por lo menos conocidos, aunque el antecedente de
diverticulosis aumenta la posibilidad de diverticulitis.
El recuento de
glóbulos blancos era de 16100/mm3 con 87 por ciento de neutrófilos y 7 por
ciento de formas en banda. El nivel de hemoglobina era de 12,7 g/dl con un
volumen corpuscular medio normal. Las plaquetas eran de 85000/mm3. La
creatinina sérica era de 1,4 mg/dl, y el nivel de urea era de 34 mg/dl. Los
electrolitos eran normales, y el bicarbonato era de 27 mmol/litro. El tiempo de
protrombina estaba prolongado a 16,2 seg con un RIN de 1,8, y el tiempo parcial
de tromboplastina era normal de 31 segundos. La eritrosedimentación era de 57mm
en la hora. Los niveles de amilasa y lipasa eran normales y el resultado de los
tests de función hepática eran normales excepto una albúmina de 2,6 g/dl. No se
obtuvo un frotis de sangre periférica. Los análisis de orina mostraron la presencia de sangre 3+ con 2 a 4 glóbulos
rojos y 1+ cilindro granuloso por campo de gran aumento.
La desviación a la izquierda, la elevación del recuento de glóbulos
blancos, y la eritrosedimentación reflejan una respuesta al stress o un proceso
inflamatorio o infeccioso. El nivel inicial de hemoglobina y el volumen
corpuscular medio sugieren que este proceso no es crónico y en el contexto de
una hemorragia aguda puede estar indicando que todavía no ha habido tiempo para
el equilibrio de la hemodilución. La trombocitopenia con un RIN elevado
aumenta la posibilidad de coagulación intravascular diseminada en el contexto
de infección intraabdominal o cáncer. Se requiere en este punto un panel de
tests dirigidos a descartar coagulación intravascular diseminada. Dado que el
paciente tiene hipoalbuminemia, el RIN elevado puede indicar insuficiencia
hepática.
El nivel elevado de urea y creatinina y los cilindros granulares en el
análisis de orina son compatibles con pérdida de volumen, aunque la pérdida de
sangre gastrointestinal es también probable que haya contribuido a la elevación
del nitrógeno ureico en sangre. Dado el pequeño número de glóbulos rojos vistos
en el sedimento urinario y el test francamente positivo con la tira reactiva
para sangre, existe la posibilidad de mioglobinuria quizás secundaria a
miositis isquémica de la pierna.
Una radiografía de
tórax no mostró enfermedad aguda, una radiografía de abdomen no mostró
obstrucción, ni aire libre ni cálculos. Una TC de abdomen y pelvis reveló un
área de diverticulosis sigmoide (Figura 1), engrosamiento de la pared del colon
sigmoide, y una arteria ilíaca derecha calcificada con infiltración de la
grasa adyacente (Figura 2).
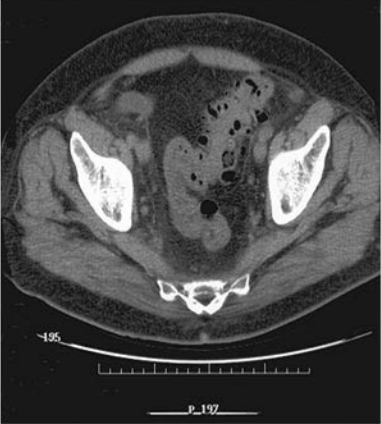
Figura 1.
TC de pelvis
obtenida sin la administración de material de contraste que muestra extensa diverticulosis
sigmoidea.

Figura 2.
TC de pelvis
mostrandouna arteria ilíaca calcificada en su periferia (A) con infiltración de
la grasa adyacente (B).
El examen de la radiografía simple es útil para descartar perforación
libre y obstrucción. La TC muestra hallazgos inespecíficos y es compatible con
la presencia de diverticulitis, colitis isquémica o enfermedad inflamatoria
intestinal. La presencia de sangrado gastrointestinal bajo no es una
observación frecuente en el curso de una diverticulitis. Una fístula
arterioentérica sigue siendo motivo de preocupación. Una colonoscopía podría
ser útil pero no fue llevada a cabo presumiblemente por el intenso sangrado y
la falta de preparación colónica, así como por la posibilidad de empeorar una
diverticulitis aguda.
Se obtuvieron
muestras para hemocultivos, y se comenzó con terapia empírica con
levofloxacina, metronidazol, y gentamicina, así como aporte vigoroso de
líquidos por vía intravenosa. Hubo un mejoramiento hemodinámico inicial pero el
paciente presentó un nuevo episodio de hematoquecia seguido de síncope con
caída de la presión arterial a 69/41 mm Hg. Un segundo análisis de sangre
obtenido varias horas después del inicial mostró un nivel de hemoglobina que
había caído a 7,6 g/dl, que las plaquetas habían caído a 55.000 por mm3. El
dolor en la pierna derecha empeoró y el pie derecho estaba más frío que el
izquierdo. Se consultó a un cirujano general y a un cirujano vascular.


Cuál es el diagnóstico?
La resolución del caso se publicará el domingo 07/04/2013
El paciente está desarrollando una insuficiencia arterial. Un Doppler
dúplex arterial puede mostrar un trombo, una compresión o un aneurisma. Dado
que los hallazgos de la TC son intrascendentes desde el punto de vista
vascular, sería útil una angiografía, ya que pequeñas fístulas o erosiones
arteriales pueden ser no detectadas por ultrasonografía. Sin embargo, cuando la
función renal está comprometida, habría que utilizar un centelleograma como
método alternativo en la detección de hemorragia. A veces es necesaria una
cirugía abierta para establecer un diagnóstico definitivo de erosiones o
fístulas arteriales. En este caso, el paciente requiere antes de la cirugía una
adecuada resucitación con líquidos. Una vez que la condición del paciente se
estabilice, un estudio para localizar el sitio de la hemorragia debe ser
realizado inmediatamente; hay una ventana de 6 a 10 horas antes de que se
inicie un proceso isquémico irreversible.
Si la intervención de cirugía vascular es necesaria debería agregarse
una cobertura antibiótica para gram positivos dado la probable contaminación
por estafilococos de piel. También debe apuntalarse el aspecto hematológico del
paciente incluyendo transfusión de plasma fresco congelado, plaquetas y
glóbulos rojos.
Se le administró
glóbulos rojos, plasma fresco congelado y plaquetas. La presión sanguínea
aumentó a 134/80 mm Hg, pero el tiempo de protrombina permaneció elevado. Un
ultrasonido dupplex no pudo demostrar flujo en la arteria femoral común derecha
(Figura 3) y ausencia de presión sistólica en el tobillo derecho. Un scan con
tecnecio-99m (Figura 4) demostró sangrado activo en colon rectosigmoideo.

Figura 3.
Ultrasonografía
Doppler de la arteria femoral común derecha demuestra ausencia de flujo en
ambos colores (flecha), y una velocidad y forma que indica ausencia de flujo
vascular (cabeza de flecha)

Figura 4.
Centelleograma con
glóbulos rojos marcados con Tecnecio 99 que muestra una actividad del trazador
localizada en el lado derecho de la pelvis indicando hemorragia en la unión
rectosigmoidea.
El scan confirmó el
sitio del sangrado a nivel del colon sigmoide con una aparente fístula
arterioentérica; esta es una condición que pone potencialmente en peligro la
vida del paciente. Una diverticulitis perforando la arteria ilíaca podría
explicar todos los síntomas del paciente aunque esta es una rara complicación
de una diverticulitis y cuando ocurren son más frecuentes en el lado izquierdo.
Se requiere cirugía de emergencia.
El paciente fue
llevado a la sala de cirugía. La laparotomía reveló diverticulosis y extensa
calcificación de la aorta y los vasos ilíacos. La exploración reveló un cuerpo
extraño sólido perforando la unión rectosigmoidea a nivel de la arteria ilíaca
externa derecha, que producía extensa hemorragia y trombo en el colon y en los
vasos. Una arteriotomía y una tromboembolectomía fueron llevadas a cabo pero la
contaminación fecal del campo hizo que no fuera posible la colocación de un
injerto vascular durante la reparación arterial. Se cerró la laceración ilíaca; un estudio
Doppler intraoperatorio mostró una tenue señal distal a la reparación sugiriendo
un mínimo mejoramiento en el flujo vascular. Una colectomía sigmoidea con
colostomía fue llevada a cabo sin complicaciones. En los hemocultivos
desarrollaron Clostridium ramosum y C. cadaveris. El examen anatomopatológico
reveló cambios inflamatorios, diverticulosis con un divertículo perforado por
un escarbadientes de 5,8 cm por 0,3 cm. El postoperatorio se complicó con dolor
e isquemia persistente de la pierna derecha. Procedimientos de
revascularización adicionales no se llevaron a cabo debido a la posibilidad de infección y daño isquémico
irreversible. El desarrollo de gangrena terminó con amputación a nivel de
rodilla en el sexto día de postoperatorio y una amputación por encima de la
rodilla 20 días más tarde por mala cicatrización de la herida operatoria del
muñón. El paciente no recordaba haber ingerido o masticado un
escarbadientes.
Comentario
Las perforaciones
del tracto gastrointestinal por escarbadientes ingeridos son raras con una tasa
anual de 0,2 por 100.000 personas. (1) De las ingestiones de cuerpos extraños
sin embargo, las reportadas por escarbadientes han mostrado la mayor tasa de
impactación y perforación (9 por ciento). (2) Esos pequeños, finos e
indigeribles cuerpos extraños tienen al menos una punta terminada en forma muy
punzante y su largo los hace dificultosos de atravesar la tortuosa luz
intestinal. (3) Las perforaciones ocurridas a lo largo del tracto
gastrointestinal, incluyendo el duodeno, yeyuno, ciego, sigmoide, apéndice y
divertículo de Meckel, (4) con complicaciones que incluyen formación de
abscesos, sepsis, hemorragia, y perforaciones de vasos mayores que resultan en
muerte. (5,6,7,8,9) Este paciente tuvo una complicación arterial mayor
complicada con sepsis y gangrena de miembro inferior que requirió amputación.
La oportuna identificación y tratamiento de la perforación por el escarbadiente
fue requerida para que el paciente no muriera a causa de hemorragia masiva y
sepsis. Sin embargo, el diagnóstico permanece siendo un desafío ya que los pacientes a veces no recuerdan
haber ingerido un escarbadientes, y los síntomas suelen confundir con otros
muchos diagnósticos. Un escarbadientes no es visible radiográficamente.
Este paciente no
recordaba haber ingerido o masticado un escarbadientes, y esta respuesta no es
un fenómeno infrecuente. En una serie de pacientes en los que se encontró
ingestión de escarbadientes sólo 12 por ciento recordaban que el episodio
hubiese ocurrido. (10) Muchos pacientes afectados tenían alterada la
sensibilidad oral o tenían prótesis, usaban alcohol, o tenían alteraciones
psiquiátricas de base. (1) La ingestión accidental ha sido también reportada en
adultos competentes especialmente en aquellos que habitualmente mastican
escarbadientes, aunque este paciente negó tal práctica. (9) El uso de dentadura
fue su principal factor de riesgo.
Los síntomas y
signos asociados con la perforación por escarbadientes son similares a otras
muchas enfermedades intraabdominales incluyendo diverticulitis, apendicitis,
cólicos renales y enfermedad inflamatoria intestinal. (10,11,12) Dado los
antecedentes de enfermedad diverticular en este paciente y los síntomas de
anorexia, malestar y náuseas, los clínicos inicialmente sospecharon
diverticulitis. El agregado de isquemia en la pierna derecha y el sangrado
rectal al dolor abdominal fueron los datos claves para la sospecha de fístula
arterioentérica dado que este diagnóstico proveía una explicación unificadora a
esta forma de presentación clínica tan
inusual de un trastorno intraabdominal. Aunque en la cirugía no permitió identificar claramente la causa de la
hematuria, esta puede haber estado relacionada a inflamación ureteral.
Dado que el
escarbadientes es radiolúcido fue difícil detectarlo en las imágenes. En una
serie de 57 pacientes que ingirieron escarbadientes esos cuerpos extraños
fueron visualizados por ultrasonografía, TC y radiografías en sólo 14 por
ciento. (10)
En el caso bajo
discusión, la intensidad del sangrado y otros factores contraindicaban la
colonoscopía. Aunque la sensibilidad y especificidad del scan nuclear no son de
una sensibilidad y especificidad óptima (85 por ciento y 70 por ciento
respectivamente), (13) en este caso la técnica nuclear identificó el sitio de
sangrado permitiendo una pronta y definitiva intervención quirúrgica. La
laparotomía confirmó la perforación del colon sigmoide por un escarbadientes
con penetración en la arteria ilíaca externa derecha.
La diverticulosis
colónica es un factor predisponente para perforación. (14) Dado la evidencia de
perforación diverticular en el examen anatomopatológico, es probable que el
escarbadientes pueda haberse alojado en un divertículo sigmoideo y haya sido
forzado por la peristalsis a un giro agudo a la derecha (“sharp right turn”) lo
que resultó en penetración de la mucosa y de la arteria ilíaca externa, y
finalmente una fístula arterial. La desafortunada combinación de enfermedad
vascular preexistente con trauma, hemorragia, y subsecuente infección
resultaron en cambios isquémicos que llevaron a la gangrena y la amputación del miembro inferior comprometido.
CONCLUSIONES DEL
CASO
Hematoquecia,
seguida de síncope y dolor en pierna derecha fue la forma de presentación de
este hombre de 60 años. La hematuria asociada inicialmente fue un elemento que
agregó confusión al cuadro, que no se repitió y que finalmente no fue explicado
su mecanismo ni aun durante el
procedimiento quirúrgico. Se ensayó un intento explicativo con el planteo de
probable “inflamación ureteral”. Sin embargo el dato de la hematuria,
probablemente haya sido útil para los
médicos que atendieron al paciente en la localización anatómica del sitio de
sangrado, ya que hematoquecia no es sinónimo de hemorragia digestiva baja, y
algunos sangrados por encima del ángulo de Treitz con un acelerado tránsito
intestinal pueden cursar con la emisión de sangre roja por vía rectal.
El compromiso
hemodinámico asociado a síncope e hipotensión ortostática como en este caso
sugieren hemorragia masiva de más de 1000 ml. Las medidas de resucitación,
expansión con cristaloides y soporte hemodinámico no estabilizaron
completamente al paciente, y un segundo episodio de sangrado agregó
inestabilidad al cuadro lo que finalmente precipitó la laparotomía exploradora como medida heroica diagnóstica y terapéutica. La presencia de dolor severo en
miembro inferior de este paciente en shock, son sugestivos de isquemia crítica
a dicho nivel. Esta isquemia puede ser causada por trombo arterial, o bien ser
secundaria al cuadro hemodinámico sistémico de hipotensión crítica, asociado a
anemia aguda, actuando sobre un
territorio vascular previamente enfermo. En este caso se dieron ambas cosas a
lo que se sumó un probable fenómeno de “robo” ocasionado por una fístula
arterio-entérica.
La mayoría de las hemorragia digestivas bajas masivas en pacientes de este grupo etario son producidas
por diverticulosis colónica, alteraciones vasculares (angiodisplasias, colitis
isquémicas, colitis por radiación), causas inflamatorias (enfermedad
inflamatoria intestinal o enteritis infecciosas), y neoplasias. Las fístulas
arterio-entéricas son causa rara de hemorragia digestiva pero deben ser siempre
consideradas en el espectro de diagnósticos diferenciales en los sangrados
masivos, para no dejar pasar la oportunidad del diagnóstico inmediato de este
cuadro potencialmente mortal. Las comunicaciones entre el árbol vascular y el
tubo digestivo son el resultado de fístulas entre la aorta y la tercera o
cuarta porción del duodeno como localización más frecuente, seguidas por el
yeyuno y el íleon, y son generalmente consecuencia de un aneurisma de aorta
abdominal aterosclerótico complicado, o de una prótesis vascular colocada
previamente en posición aórtica que se necrosa por presión o por
infección. Fístulas arterio-entéricas
más bajas como sucedió en este caso,
entre el colon y la arteria ilíaca derecha son la mayor parte de las
veces consecuencia de radioterapia previa, complicaciones de prótesis aórticas,
complicaciones vasculares de riñones trasplantados o la ingestión de cuerpos
extraños como sucedió en este caso. Las fístulas arterio-entéricas son cuadros
frecuentemente cataclísmicos, cuya forma
de presentación es hematoquecia masiva y sepsis. Las fístulas ilíaco-entéricas
son un subgrupo de fístulas entero-vasculares que como todo este grupo de trastornos
requieren un alto índice de sospecha diagnóstica, una resucitación agresiva y
un tratamiento quirúrgico inmediato para salvar la vida del paciente.
Finalmente digamos
que entre las fístulas entero-vasculares por ingestión de cuerpo extraño, la
producida por ingestión de escarbadientes está ampliamente documentada en la
bibliografía, observándose principalmente en pacientes desdentados con prótesis
dentarias o en aquellos que tienen el hábito de masticar estos elementos. A
veces los pacientes no recuerdan haberlos ingerido, lo que asociado a la
radiolucidez de los escarbadientes hace difícil la sospecha del diagnóstico el
cual es establecido a mayoría de las veces durante la cirugía.
Fuente.
From the Department of Medicine (A.K.M., M.T.F.) and the Divisions of
Vascular Surgery (B.L.J.) and Gastroenterology (P.G.B.), University of South
Florida College of Medicine, Tampa.
Address reprint requests to Dr. Flannery at the University of South
Florida College of Medicine, Department of Internal Medicine, 4 Columbia Dr.,
Suite 630, Tampa, FL 33606, or at robert_k_mohanty{at}yahoo.com .
Referencias
1. Budnick LD.
Toothpick-related injuries in the United States, 1979 through 1982. JAMA
1984;252:796-797. [Abstract]
2. McManus JE. Perforation
of the intestine by ingested foreign bodies. Am J Surg 1941;53:393-402.
3. Schwartz JT, Graham DY.
Toothpick perforation of the intestines. Ann Surg 1977;185:64-66.
[ISI][Medline]
4. Callon RA Jr, Brady PG. Toothpick
perforation of the sigmoid colon: an unusual case associated with
Erysipelothrix rhusiopathiae septicemia. Gastrointest Endosc 1990;36:141-143.
[ISI][Medline]
5. Cockerill FR III, Wilson
WR, Van Scoy RE. Traveling toothpicks. Mayo Clin Proc 1983;58:613-616.
[ISI][Medline]
6. Monkemuller KE, Patil R,
Marino CR. Endoscopic removal of a toothpick from the transverse colon. Am J
Gastroenterol 1996;91:2438-2439. [ISI][Medline]
7. Justiniani FR, Wigoda L, Ortega RS. Duodenocaval
fistula due to toothpick perforation. JAMA 1974;227:788-789.
[CrossRef][ISI][Medline]
8. Dicicco BS, Heit HA,
Peterson JE, Harshaw WG, Cooper JN. Massive bleeding due to arterial-enteric fistula
from an ingested toothpick. J Clin Gastroenterol 1985;7:292-295. [ISI][Medline]
9. Bee DM, Citron M, Vannix
RS, et al. Delayed death from ingestion of a toothpick. N Engl J Med
1989;320:673-673. [ISI][Medline]
10. Li SF, Ender K. Toothpick
injury mimicking renal colic: case report and systematic review. J Emerg Med
2002;23:35-38. [CrossRef][ISI][Medline]
11. Tenner S, Wong RC,
Carr-Locke D, Davis SK, Farraye FA. Toothpick ingestion as a cause of acute and
chronic duodenal inflammation. Am J Gastroenterol 1996;91:1860-1862.
[ISI][Medline]
12. Guber MD, Suarez CA,
Greve J. Toothpick perforation of the intestine diagnosed by a small bowel
series. Am J Gastroenterol 1996;91:789-791. [ISI][Medline]
13. Rantis PC Jr, Harford FJ,
Wagner RH, Henkin RE. Technetium-labelled red blood cell scintigraphy: is it
useful in acute lower gastrointestinal bleeding? Int J Colorectal Dis
1995;10:210-215. [CrossRef][ISI][Medline]
14. Carp L. Foreign bodies in
the intestine. Ann Surg
1927;85:575-591.