Una mujer de 20 años fue internada en el hospital en su semana 26 de gestación por mareos, confusión y dificultades en la marcha.
Diez semanas antes de la internación, la paciente se realizó un test casero de embarazo que fue positivo, consultando a un centro de salud para control de su embarazo. Un test para drepanocitosis, sífilis, HIV, hepatitis B y C fueron negativos. Tests serológicos de IgG para virus varicela-zoster y rubeola fueron positivos. Dos semanas más tarde, una muestra endocervical fue positiva para infección por Chlamydia trachomatis, y negativa para gonorrea. La paciente perdió el turno para consulta y comenzó el tratamiento con azitromicina 4 semanas más tarde.
Seis semanas antes de la internación, se mudó a un asilo para mujeres embarazadas. Los miembros del staff del asilo la describieron como feliz, presentando conducta aniñada, mala memoria, confusión, y movimientos extraños con su cabeza. Durante las siguientes 2 semanas, aparecieron diariamente náuseas y vómitos, que fueron controlados con metoclopramida. Cuatro días antes de la internación,desarrolló mareos y pérdida de fuerzas en el lado izquierdo del cuerpo; comenzó a presentar caídas hacia el lado izquierdo y a presentar vómitos varias veces en el día. Al día siguiente, concurrió a un departamento de emergencias.
En la evaluación en emergencias, la paciente estaba orientada en espacio pero no en tiempo, día ni mes, y brindó datos contradictorios acerca de sus antecedentes médicos. El útero era grávido, y el resto del examen físico era normal. Un electrocardiograma reveló taquicardia sinusal y rotación antihoraria del eje de la onda T en precordiales. Los análisis de orina mostraron proteinuria de 30 mg/dl y un nivel de glucemia de 100 mg/dl. En el screening toxicológico existía positividad para metabolitos tricíclicos en orina.
Una TAC de cerebro reveló una leve prominencia difusa del sistema ventricular. No había masas ni otras lesiones focales.
En el segundo día de internación, la paciente estaba alerta, tranquila, cooperativa, y orientada en persona, lugar, y eventos corrientes, aunque no estaba conciente acerca de detalles de su vida. No había temblor, ni signos extrapiramidales. El examen ultrasonográfico reveló una anatomía y crecimiento fetal normales, correspondiente a una gesta de 25 semanas y 6 días. En el tercer día de internación, la debilidad, náuseas y vómitos se resolvieron, y se consideró que había vuelto a su status mental normal. Fue dada de alta al asilo con recomendaciones de seguimiento por consultorio externo de neurología. En el asilo, la paciente volvió a estar mareada, con dificultad para caminar, presentando un episodio de caída de la silla. Esa tarde fue llevada nuevamente al departamento de emergencias.
En el departamento de emergencias, la paciente reportó sentirse “atontada” y nauseosa. Se quejaba de cefalea leve de reciente comienzo, en localización en banda a nivel de la ceja. El interrogatorio mostraba contradicciones; la historia clínica fue provista por los miembros del staff del asilo, pero muchos detalles no pudieron obtenerse. La paciente era nativa de Cabo Verde en el Atlántico Africano, y había migrado a los EE UU hacía 3 años. Había tenido sarampión a los 4 meses de edad y varicela en su niñez. A los 7 años de edad, presentó traumatismo de cráneo en una caída pero dijo haberse recuperado totalmente. Las vacunas, incluyendo la de poliomielitis, difteria, pertusis, y tétanos estaban completas. La vacuna del sarampión la había recibido a los 11 meses; posteriormente, vacunas combinadas de sarampión, parotiditis, y rubeola; y la vacuna contra hepatitis B, 2 años antes de entrar en la escuela secundaria.
Estaba desempleada. Los últimos 3 años había vivido con parientes, amigos, y un novio, así como en asilos. Era soltera y no tenía relación social con el padre de su feto. Sus padres y siete hermanas estaban vivas pero no estaban en contacto con ella. No había historia familiar disponible. No presentaba alergias conocidas, no tomaba alcohol, tabaco ni drogas ilícitas.
En el examen en el departamento de emergencias, la paciente estaba alerta pero no colaboraba, y presentaba movimientos cefálicos involuntarios. Su status mental, según un amigo era el habitual. La presión arterial era de 107/81 mmHg, el pulso de 84 por minuto, y la temperatura de 36,3ºC; la frecuencia respiratoria de 18 por minuto, y la saturación de oxígeno de 100% mientras respiraba aire ambiente. Tenía lesiones acneiformes en la cara. El abdomen era blando, grávido, y no dolía; el feto impresionaba sano. El primer par craneal no fue evaluado, pero del 2º al 12º eran normales. La fuerza era normal, y la marcha insegura. El resto del examen era normal.
Los resultados de laboratorio se muestran en la Tabla 1. Después de premedicación con lorazepan a una dosis de 1 mg para controlar los movimientos involuntarios, se realizó una RMN cerebral de cerebro sin administración de sustancia de contraste. En imágenes en T2, y FLAIR (fluid-attenuated inversion recovery), se vieron señales hiperintensas en hipocampo y giro parahipocámpico izquierdos, así como en la región posterior de la cápsula interna izquierda. No había evidencias de alteraciones de la difusión.
 Tabla 1. Resultados de los Tests de Laboratorio.
El examen realizado por un neurólogo interconsultado mostró que la paciente estaba orientada en persona y lugar, con conductas infantiles. Su lenguaje era fluido, y la nominación estaba intacta. Podía leer una frase corta, Era zurda, podía escribir su nombre pero no una oración, y podía seguir órdenes simples. Su atención era variable y los tests de memoria mostraron mostraron recuperación de cero de tres items a los 5 minutos en la repetición del examen. Había leve asimetría de la cara con pérdida del pliegue nasolabial derecho. El olfato y el gusto no fueron examinados, pero el resto de los pares craneales eran normales. Había movimientos coreiformes de la cabeza y cuello, podía llevar a cabo con dificultad movimientos rápidos alternantes, y había apraxia. Había hipertonía e hiperreflexia con clonus en la pierna derecha. Había aumento de la base de sustentación en la marcha, con inestabilidad postural e inclinación hacia la izquierda. Era incapaz de mantenerse parada en un pie. Fue admitida al servicio de neurología.
Tabla 1. Resultados de los Tests de Laboratorio.
El examen realizado por un neurólogo interconsultado mostró que la paciente estaba orientada en persona y lugar, con conductas infantiles. Su lenguaje era fluido, y la nominación estaba intacta. Podía leer una frase corta, Era zurda, podía escribir su nombre pero no una oración, y podía seguir órdenes simples. Su atención era variable y los tests de memoria mostraron mostraron recuperación de cero de tres items a los 5 minutos en la repetición del examen. Había leve asimetría de la cara con pérdida del pliegue nasolabial derecho. El olfato y el gusto no fueron examinados, pero el resto de los pares craneales eran normales. Había movimientos coreiformes de la cabeza y cuello, podía llevar a cabo con dificultad movimientos rápidos alternantes, y había apraxia. Había hipertonía e hiperreflexia con clonus en la pierna derecha. Había aumento de la base de sustentación en la marcha, con inestabilidad postural e inclinación hacia la izquierda. Era incapaz de mantenerse parada en un pie. Fue admitida al servicio de neurología.
El segundo día de internación se llevó a cabo una punción lumbar. Los resultados de la punción del líquido cefalorraquídeo se muestran en la Tabla 2; otros resultados se muestran en la Tabla 1. Un ELISA para HIV fue negativo. Un electroencefalograma mostró enlentecimiento difuso de ondas tetha, y una actividad delta rítmica intermitente frontal, que era más prominente en el hemisferio derecho que en el izquierdo. No había actividad epileptiforme (Figura 1). Se repitió una RMN de cerebro después de la administración de gadolinio que no mostró evidencias de realce anormal. Se administró aciclovir por vía intravenosa.

Tabla 2. Resultados de los Tests del Líquido Cefalorraquídeo.

Figura 1. Electroencefalograma Obtenido en el Segundo Día de Hospital.
El EEG es marcadamente anormal, mostrando modesto enlentecimiento del ritmo de base posterior dominante, con salvas de actividad delta rítmica intermitente (flechas), a veces máxima en el lado derecho y otras veces relativamente simétricas bilateralmente. No había elementos epileptiformes ni descargas periódicas.
Al día siguiente, un test sérico para enfermedad de Lyme, un hisopado faríngeo para ácidos nucleicos de Mycoplasma pneumoniae, y cultivos de sangre y orina fueron todos negativos; los resultados de otros tests son listados en la Tabla 1. El quinto día de internación, la condición de la paciente pareció mejorar. Estaba orientada y recordaba detalles de su pasado; la dismetría y la ataxia de tronco se habían reducido. Un nuevo electroencefalograma no mostró cambios. Al día siguiente, se llevó a cabo otra punción lumbar (Tabla 2).
Entre el 7º y el 18º días de hospital, la función motora de la paciente empeoró gradualmente, desarrolló una sensación de que el lado derecho de su cuerpo no le pertenecía, se volvió incapaz de alimentarse por si misma, su capacidad de respuesta y la de seguir órdenes simples disminuyeron, y apareció incontinencia.. Comenzó a adoptar una posición fetal, a emitir gemidos y llantos con gritos ininteligibles. Un test cutáneo para tuberculosis, un hisopado nasofaríngeo en busca de antígenos respiratorios virales y cultivos virales de una muestra de materia fecal fueron todos negativos. Una carga viral para RNA de HIV, así como un test de anticuerpos dirigidos contra HIV fueron negativos. Los niveles de tiroxina total y libre fueron normales, y el nivel de tiroglobulina era elevado, 54,7 ng/ml (normal 4 a 40). En el día 12º, se discontinuó el aciclovir, y se comenzó con ceftriaxona 2 grs EV. Un nuevo estudio electroencefalográfico mostró atenuación aumentada de la actividad de base, y actividad rítmica intermitente frontal delta menos abundante. La RMN en el 13º día, mostró nuevas señales hiperintensas en protuberancia y pedúnculos cerebelosos medios con restricción de difusión de agua asociada en T2 y FLAIR. La restricción de la difusión fue notada en el brazo posterior izquierdo de la cápsula interna izquierda. Había atrofia en el lóbulo temporal medio izquierdo, con resolución de las señales hiperintensas anormales en las imágenes en FLAIR. En el día 14º, una tercera punción lumbar fue llevada a cabo.
Diagnóstico Diferencial:
La paciente vivía semi-independientemente hasta su embarazo, 27 semanas antes de su internación. Su nivel de función cognitiva en ese momento era desconocido, y dado la carencia de información, no fue posible determinar si la afectación de su función eran previas o secundarias a su enfermedad.
El diagnóstico diferencial neurológico se basa primeramente en el examen físico para localizar las lesiones y en el interrogatorio, especialmente la forma de comienzo, y el ritmo de la progresión, para identificar el proceso patológico. El examen neurológico de la paciente mostró una función cognitiva anormal, indicando disfunción de la sustancia gris cortical y subcortical, función motora anormal, indicando disfunción del sistema motor piramidal, y movimientos coreiformes, indicativos de disfunción del sistema motor extrapiramidal. El examen también mostró una marcha torpe, y dificultad en llevar a cabo movimientos alternantes rápidos, indicando disfunción del cerebelo o de sus conecciones. Al no tener información del estado previo de la paciente, no tenemos información del ritmo de instalación de la enfermedad, de su “tempo”, y por lo tanto, necesitamos considerar enfermedades congénitas, y adquiridas que pueden ocasionar evoluciones agudas, subagudas, o crónicas, con episodios de estabilización, o evolución progresiva.
Nosotros hemos basado nuestros diagnósticos diferenciales en el examen neurológico, en los tests iniciales de laboratorio, y en los hallazgos de EEG y RMN.
Examen del Líquido Cefalorraquídeo.
Los resultados del análisis del líquido cefalorraquídeo en esta paciente, mostró una pleocitosis linfocítica, con pocos glóbulos rojos, un nivel levemente elevado de proteínas, y un nivel de glucosa normal. Estos hallazgos son característicos de meningitis aséptica. Por eso nosotros estábamos preocupados por la posibilidad de infecciones por virus, ricketsias, espiroquetas, infecciones decapitadas (parcialmente tratadas), focos parameníngeos de infección, ciertas enfermedades autoinmunes tales como lupus eritematoso sistémico o enfermedad de Behçet, vasculitis, carcinoma, reacciones a los efectos tóxicos de ciertas medicaciones tales como AINES, y meningitis químicas relacionadas con la ruptura de un quiste. Aunque inespecíficos, los hallazgos del líquido cefalorraquídeo proveen sostén a la posibilidad de infección aguda o subaguda, o enfermedad inflamatoria. La presencia de respuesta inflamatoria hace que enfermedades crónicas como enfermedad de Huntington, enfermedad de Wilson, y abiotrofias sistémicas tales como atrofias multisistémicas sean improbables.
Estudios Electroencefalográficos.
El EEG inicial era marcadamente anormal, pero los hallazgos fueron inespecíficos (Figura 1). El ritmo lento dominante y la atenuación posterior sugieren enfermedad de la sustancia gris cortical, mientras que la actividad delta frontal rítmica intermitente sugiere enfermedad de sustancia gris subcortical. Las ondas lentas monomórficas también sugieren que inicialmente la sustancia blanca subcortical estaba relativamente respetada. Dr Henson, puede revisar los estudios radiológicos?
Las imágenes axiales en T2 y FLAIR en la RMN de cerebro el día de la internación, llevadas a cabo sin administración de gadolinio, reveló una región de señal hiperintensa en el lóbulo temporal medio izquierdo (Figura 2A) y un sutil aumento de la señal en el brazo posterior de la cápsula interna izquierda, correspondiente a la localización del tracto corticoespinal. Esos focos no mostraron restricción de la difusión o realce anormal en el estudio llevado a cabo con gadolinio al día siguiente. El aspecto de la protuberancia no era remarcable, y no se notaron otros hallazgos significativos. La venografía por RMN de cerebro fuen normal.

Figura 2. Estudio de Imágenes Cerebrales.
Una RMN en T2 con FLAIR (Panel A), obtenida el día de la internación mostró una región anormal de señal hiperintensa en el hipocampo izquierdo (flecha) y en el brazo posterior de la cápsula interna (no mostrado). El día 13º de internación existía una nueva región de señal hiperintensa en T2 FLAIR en protuberancia y pedúnculos cerebelosos medios (Panel B flechas), y restricción de la difusión en pedúnculos cerebelosos medios, y brazo posterior de la cápsula interna como se muestra en las imágenes de difusión (Panel C, flechas).
Alrededor del día 13º de internación existían marcados cambios. Había una zona de señal anormal en la protuberancia (Figura 2 B), con áreas de difusión restringida. La hiperintensidaddel lóbulo temporal izquierdo medio se había resuelto, y había pérdida de volumen en la región del hipocampo. Había difusión restringida en el tracto corticospinal (Figura 2 C); no se detectaron realces anormales. Estos hallazgos fueron interpretados como resultado de una encefalitis subaguda causada por una infección o un trastorno autoinmune.
En resumen, esta paciente tenía disturbios de la función cognitiva, del haz piramidal, y disfunción cerebelosa, con trastornos del movimiento consistentes en movimientos coreiformes, y tests tanto electroencefalográficos como en las imágenes sugestivos de encefalitis infecciosa o autoinmune.
TRASTORNOS DEL MOVIMIENTO.
La corea es un trastorno hiperkinético del movimiento que puede resultar de un número de enfermedades neurológicas; parece empeorar durante el embarazo, condición conocida como corea gravidarum, o corea gravídica. La mayoría de las pacientes con esta condición, se presentan durante el segundo trimestre del embarazo con movimientos aislados, que se resuelven después del parto. Las causas más comunes son la fiebre reumática aguda (corea de Sydenham), y el síndrome antifosfolipídico. Nosotros también consideramos otras enfermedades asociadas con corea como el lupus eritematoso sistémico, la corea de Huntington, y la enfermedad de Wilson, aunque las últimas dos enfermedades fueron descartadas por los resultados del líquido cefalorraquídeo y por otros elementos.
La corea de Sydenham es una complicación tardía de la infección con estreptococo del grupo A; el comienzo ocurre meses después de la infección aguda. La mayoría de los casos ocurre en la niñez, pero hasta el 30% de los pacientes pueden tener corea recurrente meses o años después del episodio inicial. (1) Esta paciente tuvo sólo un anticuerpo antiestreptolisina mínimamente elevado, sin ninguna otra evidencia de infección o enfermedad cardíaca. El síndrome antifosfolipídico (2) puede ser primario o secundario a lupus eritematoso sistémico, y puede comenzar a manifestarse durante el embarazo. La paciente no tuvo síntomas ni signos de lupus sistémico, pero el lupus confinado al sistema nervioso nervioso central es una entidad perfectamente descripta, y puede empeorar durante el transcurso del embarazo. Los tests para anticuerpos antifosfolípidos fueron todos negativos, así como los tests para lupus.
ENCEFALITIS VIRAL AGUDA.
La encefalitis por herpes simplex fue inicialmente considerada como causa de encefalitis aguda infecciosa en esta paciente dado la anomalía detectada en el hipocampo izquierdo en la RMN. A diferencia de las encefalitis arbovirales y la encefalitis del Oeste del Nilo, que ocurren en el verano y otoño, cuando los mosquitos son abundantes, la encefalitis por herpes simplex ocurre esporádicamente a lo largo de cualquier parte del año. Los resultados negativos de los tests para ácidos nucleicos para virus de herpes simplex en LCR, y la falta de respuesta al aciclovir, hizo que se considerara a este diagnóstico como improbable. Otras causas comunes de encefalitis virales incluyen infecciones por enterovirus y echovirus, así como infecciones por coxscakievirus, que típicamente producen signos prominentes de irritación meníngea, con fotofobia, meningismo, náuseas, y cefalea, pero sólo signos mínimos de disfunción cerebral focal, que pueden ser fugaces y no progresivos. Los tests serológicos en esta paciente, descartaron a estos agentes también como causa.
ENCEFALITIS SUBAGUDAS.
Encefalitis Paraneoplásicas.
La encefalitis límbica paraneoplásica puede preceder a la aparición de un tumor por meses o aún años. Síntomas psiquiátricos, alteraciones en la memoria, confusión y somnolencia, trastornos respiratorios, ataxia, y parálisis de nervios craneales han sido reportados. Hay un modesto aumento de las proteínas en el LCR, pero escasa celularidad. La clínica de esta paciente, así como las alteraciones del LCR no eran consistentes con este diagnóstico.
Encefalitis Postinfecciosa.
Varias infecciones pueden estar asociadas consíndromes de encefalitis postinfecciosa, incluyendo sarampión, parotiditis, y rubeola, influenza, Epstein-Barr, y varicela-zoster. La encefalomielitis diseminada aguda puede ocurrir después de una variedad de infecciones virales y después de la aplicación de la vacuna de la rabia y de la viruela. (3) Usualmente comienza con síntomas inespecíficos tales como fiebre, cefalea, rigidez de nuca, vómitos, y anorexia. El examen neurológico puede mostrar neuritis óptica, ataxia, y signos de foco neurológicos motores; convulsiones y alteraciones de conciencia pueden ocurrir. Esta paciente no tenía antecedentes de inmunizaciones recientes, infecciones virales, o evidencias de neuritis óptica, y las imágenes no eran las típicas vistas en la encefalomielitis diseminada aguda.
El LCR fue una importante pista diagnóstica en este caso. Aunque el nivel inicial de proteínas en LCR estaba levemente aumentado, el componente IgG estaba marcadamente elevado. Dr. Roehrl, podría discutir la importancia de este hallazgo?
El nivel de proteínas totales en LCR, y las IgG, obtenidas el 2º día de internación se muestran en la Tabla 2. La electroforesis en gel de agarosa del LCR reveló bandas oligoclonales en la región gama (Figura 3A). El cociente entre LCR y suero para la albúmina y las concentraciones de IgG fueron QAlb=4.7x10–3 y QIgG=26.1x10–3 , correspondientes a un índice de IgG de 5,6 (valor normal menor de 0,85) (4). En base a este análisis, desarrollado por Reiber, (5,6,7) los resultados de esta paciente (Figura 3B) indicaban marcado aumento de la síntesis intratecal de IgG (fracción de producción intratecal 87,7%) sin evidencias clínicamente significativas de disfunción de la barrera hematoencefálica. La relación de concentración de IgG sobre proteínas totales en LCR fue de 58,9%

Figura 3. Resultados de la Electroforesis del LCR.
El Panel A muestra los resultados de la electroforesis en gel de agarosa de una muestra de LCR de la paciente, obtenida el 2º día de internación (concentrado a 1/63 del volumen original) y un LCR control (concentrado 1/80 del volumen original). P, A, 1, 2, , and denotan regiones electroforéticas de prealbúmina, albúmina, alfa-1, alfa-2, beta, y gama respectivamente. La flecha muestra la posición de varias bandas densas en la región gama, indicando la presencia de múltiples bandas de inmunoglobulinas oligoclonales. Hay una disminución relativa del nivel de albúmina en el LCR de la paciente. El Panel B muestra un gráfico doble logarítmico diseñado de acuerdo al método propuesto por Reiber (también llamado Reibergrama), en el que el cociente entre la concentración en LCR de albúmina y la sérica (QAlb) así como de IgG (QIgG), son graficados en la abscisa y en la ordenada respectivamente. Los límites superiores (QHigh), e inferiores (QLow) de los valores normales son mostrados como líneas sólidas, con la línea de puntos indicando los valores promedio (QMean). Los isopercentiles (líneas discontinuas) corresponden a varias cantidades relativas de producción intratecal de IgG (20 a 80%). La línea discontinua vertical denota el límite superior del QAlb ajustado por la edad, separando la función normal (izquierda) y la función anormal (derecha) de la barrera hematoencefálica. La paciente (punteado rojo) tenía síntesis marcadamente alta de IgG intratecal (fracción de producción intratecal 87,7%) sin evidencias de disfunción significativa de la barrera hematoencefálica (zona 4). La zona 1 denota función normal, la zona 2 denota disfunción pura de la barrera hematoencefálica, la zona 3 denota una combinación de síntesis de IgG intratecal aumentada, y disfunción de la barrera hematoencefálica.
Las enfermedades que pueden provocar una respuesta intratecal de esta magnitud son la sífilis, la panencefalitis rubeólica crónica, y la panencefalitis esclerosante subaguda. (8,9,10,11) Esta paciente no tenía sífilis, como mostraron los tests serológicos y las complejas manifestaciones de esta enfermedad que no son compatibles con sífilis. La encefalitis post-rubeólica (12,13) puede afectar pacientes con antecedentes remotos o infección congénita por rubeola, presentándose como demencia progresiva, ataxia, corea, degeneración retiniana y convulsiones. El examen del LCR muestra pleocitosis, y un nivel de proteínas moderadamente elevado, con hasta un 50% de proteínas del LCR compuestas por inmunoglobulinas. El diagnóstico se confirma por un alto título de anticuerpos anti-rubeola en LCR.
Panencefalitis Esclerosante Subaguda.
El sarampión puede causar tres enfermedades distintas en el sistema nervioso central: encéfalomielitis postinfecciosa, encefalitis subaguda por sarampión, y panencefalitis esclerosante subaguda. (14)
La panencefalitis esclerosante subaguda es típicamente vista entre 7 y 10 años después de una infección por sarampión, y los pacientes se presentan comúnmente con declinación de su rendimiento escolar, cambios conductuales, cefalea, extraños movimientos espontáneos, y a veces convulsiones.(15) Los hallazgos característicos incluyen sacudidas mioclónicas que son a menudo periódicas y están asociadas con descargas epileptiformes uni o bilaterales en el electroencefalograma. Aunque la mayoría de los casos ocurren en la niñez o adolescencia, hay casos que comienzan tan tardíamente como en la quinta década de la vida. (16,17) La incidencia de panencefalitis esclerosante subaguda ha disminuido con la vacunación contra el sarampión; sin embargo, persiste en lugares donde la vacunación no es común. (18) La incidencia de esta condición está aumentada hasta 10 veces, en pacientes que adquieren sarampión antes de la edad de 2 años; esta paciente tuvo sarampión a los 4 meses de edad. La enfermedad puede presentarse durante el embarazo, posiblemente como resultado de la alteración del status inmune. (8,19)
En suma, esta paciente se presentó con una enfermedad neurológica subaguda progresiva, caracterizada por una amplia disfunción del sistema nervioso central, elementos inflamatorios en el LCR, y un extremadamente alto nivel de IgG en el mismo. Nosotros estuvimos de acuerdo en la sala con el diagnóstico de panencefalitis esclerosante subaguda como consecuencia de sarampión adquirido a los 4 meses de edad. Esta condición pudo haber sido exacerbada por el embarazo. Las muestras de suero y de LCR tomadas el 14º día de internación fueron enviadas para testear el nivel de anticuerpos contra sarampión.
El índice de anticuerpos específicos contra sarampión en LCR sobre los del suero estaban elevados a un 31,8 (valor normal menos de 1,4) (7) (Tabla 3). Los anticuerpos IgM específicos contra sarampión, no se detectaron en suero. Este perfil indica una respuesta inmune crónica contra la infeccción sarampionosa en el compartimiento del líquido cefalorraquídeo. Una comparación con los títulos de parotiditis, muestra la especificidad del proceso inmunológico. Los resultados confirman el diagnóstico de panencefalitis esclerosante subaguda en esta paciente. La enfermedad fue una consecuencia tardía de una infección persistente con virus del sarampión.

Tabla 3. Resultados de los Tests para Anticuerpos contra sarampión en Sangre y Líquido Cefalorraquídeo, Obtenidos el Día 14º de Internación.
Discusión del Manejo.
La inmunidad celular estimulando los linfocitos T helper tipo 1 (Th1), inductores de citoquinas es crucial para el clearence del virus del sarampión, la semana después de la infección, mientras que las citoquinas que inducen los linfocitos T helper tipo 2 (Th2) están involucrados en la producción de anticuerpos. Varios reportes han sugerido que el tratamiento con interferón alfa-2 intratecal (una citoquina que promueve la actividad Th1), con o sin tratamiento con el agente inmunomodulador antiviral pranobex, puede disminuir o aún detener la progresión de la panencefalitis esclerosante subaguda. (20,21,22,23,24,25) Sin embargo, no se dispone de datos definitivos que muestren la eficacia de este approach terapéutico.
Cuando hicimos el diagnóstico en esta paciente, ella tenía 28 semanas de gestación, y su condición se estaba deteriorando rápidamente. Nosotros creimos que el tratamiento estaba indicado para mejorar la evolución tanto de la madre como del feto, así que la tratamos con interferón e inosina pranobex por 8 semanas, no obteniendo ningún beneficio claro. Después del parto de un niño sano, por cesárea a las 34 semanas de gestación, se tomó la decisión de discontinuar el tratamiento, con el consentimiento de la madre de la paciente. La paciente falleció 6 semanas más tarde.
Diagnóstico: Panencefalitis Esclerosante Subaguda.
Discusión Anátomo-Patológica.
La autopsia reveló infiltrados inflamatorios conteniendo macrófagos, células plasmáticas, y linfocitos alrededor de los vasos, con destrucción neuronal y gliosis reactiva, más prominentes en tronco cerebral. (Figura 4A) No había inclusiones virales de las típicamente vistas en la encefalitis aguda por sarampión (y ocasionalmente presentes en la panencefalitis esclerosante subaguda) en núcleo o citoplasma tanto en neuronas como en células de la glía, y células endoteliales vasculares. A diferencia de otras enfermedades asociadas al sarampión, la panencefalitis esclerosante subaguda es causada por la persistencia de un tipo defectuoso de virus, que no forman partículas virales completas, pero que pueden infectar células adyacentes por contacto directo. (26,27) En la corteza cerebral, las poblaciones neuronales estaban preservadas, pero había marcada gliosis con astrocitos reactivos (Figura 4B). El tronco cerebral estaba reblandecido por el proceso patológico, pero la sustancia blanca hemisférica era más dura que lo normal; esto era evidente cuando se hacían cortes de cerebro en estado fresco. La dureza correspondía a la eclerosis de la por ese motivo llamada panencefalitis esclerosante subaguda. La sustancia blanca subcortical también mostró gliosis reactiva y extensa activación de la microglia (Figura 4C). Estos hallazgos son inespecíficos, pero junto con los estudios serológicos, son consistentes con el diagnóstico de panencefalitis esclerosante subaguda.

Figura 4. Hallazgos Cerebrales en la Autopsia.
Un marcado infiltrado inflamatorio alrededor de los vasos y destrucción neuronal son los más prominentes en estos cortes del tronco cerebral, que contiene macrófagos , células plasmáticas, y linfocitos; hay prominentes infltrados perivasculares de linfocitos (Panel A, hematoxilina-eosina). En la corteza cerebral, las poblaciones neuronales están preservadas, pero está presente una marcada gliosis (Panel B, análisis inmunohistoquímico para proteínas ácidas fibrilares). Marcada activación de la microglia está presente en la sustancia blanca subcortical con células con formas de varas CD68 positivas (Panel C, flechas; hematoxilina-eosina) (inserto, tinción inmunohistoquímica para CD68).
Las muestras fijadas y congeladas de tejido cerebral fueron enviadas al Dr. William Bellini al Centers for Disease Control and Prevention para estudios inmunohistoquímicos y moleculares. (14,28,29,30) El análisis inmunohistoquímico de antígeno nucleoproteico de virus de sarampión fue negativo. Los estudios diagnósticos moleculares, llevados a cabo para detectar porciones del genoma de virus de sarampión fue negativa en repetidos intentos de amplificación de regiones de genesde nucleoproteínas de sarampión. Débiles señales fueron detectadas en ensayos de PCR de tiempo real, pero fueron insuficientes cantidades de productos de DNA amplificado para llevar a cabo un análisis de la secuencia. Como resultado, no fue posible establecer definitivamente la presencia de virus de sarampión en este caso.
Dos años antes, la paciente había sido vista en un departamento de emergencias en otro hospital por episodios de pérdida de conciencia asociados a movimientos oculares y arqueando el dorso. En el examen presentaba movimientos involuntarios intermitentes de la cabeza y cuello, así como sacudidas en los brazos. Los reflejos osteotendinosos eran vivos y simétricos. Una TAC de cráneo y un EEG fueron normales. Se diagnosticó embarazo temprano en ese momento, y el mismo fue interrumpido por decisión de la paciente. Durante los siguientes 20 meses, la paciente fue perdida en el seguimiento médico, y no se sabe si los movimientos anormales habían continuado.
Es posible que la panencefalitis esclerosante subaguda haya comenzado con ese embarazo previo, se haya estabilizado con la interrupción del mismo, y haya empeorado con el segundo embarazo?
La panencefalitis esclerosante subaguda es casi siempre una enfermedad crónicamente progresiva, aunque están descriptos períodos de plateau en el curso clínico. La duración de la enfermedad puede ser de años, sin embargo, y retrospectivamente, sus síntomas 2 años antes pueden haber sido manifestaciones tempranas de la misma enfermedad.
Diagnóstico Anatómico:
Panencefalitis Esclerosante Subaguda, Secundaria a Infección por Virus del Sarampión.
Fuente
From the Neurology Service (A.J.C., J.W.H., M.P.F.) and the Departments of Radiology (J.W.H.) and Pathology (M.H.A.R., M.P.F.), Massachusetts General Hospital; and the Departments of Neurology (A.J.C., J.W.H.) and Pathology (M.H.A.R., M.P.F.), Harvard Medical School.
Conclusiones del Caso.
Las encefalitis responden a un número muy diverso de causas, que incluyen especialmente agentes infecciosos tales como muchos virus: herpes simplex (HSV 1 y 2), virus de la varicela (VZV), sarampión, parotiditis, rubéola, herpes virus humano tipo 6, algunos agentes bacterianos como micoplasmas, hongos, y parásitos (Toxoplasma gondii) entre otros, y también, causas no infecciosas.
Se trata de una inflamación del parénquima cerebral que puede estar afectado tanto anátomo-patológica como clínicamente en forma focal o difusa. Muchas veces puede acompañarse de signos meníngeos, en cuyo caso se las describe como meningoencefalitis.
Debe diferenciarse desde el punto de vista histológico, de la cerebritis, que es un término utilizado para describir la zona de cerebro en estadio incipiente de formación de un absceso bacteriano.
El curso de las encefalitis puede ser, dependiendo del agente causal y de las condiciones propias del paciente, agudo, subagudo y crónico. Las encefalitis agudas responden más comúnmente a etiologías virales, mientras que las formas subagudas y crónicas más comunes en la actualidad, se ven en pacientes inmunocomprometidos, y probablemente la toxoplasmosis sea el primer factor etiológico en estas dos últimas formas clínicas.
El médico debe estar atento a este tipo de entidades, especialmente al síndrome encefalítico agudo, ya que aunque la mayoría de las veces responden a causas para las que no se dispone hoy en día de tratamientos efectivos, existen dos entidades que son potencialmente tratables. Ellas son la encefalitis por herpes simplex (HSV), que es esporádica y letal tanto en neonatos como en la población sana en general, y la menos frecuente encefalitis por virus de varicela-zoster (VZV), también mortal, que se ve en inmunocomprometidos. Una identificación rápida, asociado a un tratamiento oportuno de estas dos entidades puede salvar la vida del paciente. Es por ello que muchos síndromes encefalíticos agudos son tratados empíricamente con antivirales desde el comienzo de los síntomas, a la espera de la confirmación o no de la causa.
La panencefalitis esclerosante subaguda post sarampión, es una entidad rara en la actualidad, sobre todo desde la implementación de programas de vacunación antisarampionosa obligatoria en la mayoría de los países desarrollados.
El virus del sarampión, que es un RNA virus, perteneciente al grupo de los paramixovirus, tiene un especial trofismo por el sistema nervioso central, y es así que puede afectar al mismo de tres maneras:
• Una forma aguda posinfecciosa, que se produce casi simultáneamente con el exantema, y responde a fenómenos autoinmunes.
• Una forma progresiva aguda, a la que se conoce también como encefalitis por cuerpos de inclusión, que se presenta en pacientes con algún grado de inmunocompromiso y de incapacidad de resolver efectivamente la infección. Se ve tras un corto período de latencia después del exantema.
• Una forma tardía, a veces, como en el caso que nos ocupa, remota, llamada panencefalitis esclerosante subaguda post sarampión. Esta última entidad obedece a mecanismos patofisiológicos no del todo conocidos, pero en los cuales la persistencia del virus o pequeñas secuencias genéticas del mismo (en el caso de la paciente del ateneo no se pudo aislar el virus, ni tampoco antígenos nucleoproteicos, ni una secuencia genómica lo suficientemente específica por técnicas de amplificación de DNA) en el sistema nervioso en forma latente, desencadena una reacción inmunológica dentro del sistema nervioso, caracterizada por un impresionante aumento de las IgG anti virus de sarampión. Este aumento de las IgG específicas en LCR, no tiene una correlación plasmática, lo que indica que se trata de un proceso “barrera hematoencefálica hacia adentro”. Este fenómeno inmunológico local trae como consecuencia una desmielinización preferentemente en localización perivenosa. En el caso específico de la desmielinización en esta entidad, esta se acompaña de una esclerosis (del griego sklerós o “duro”) de la sustancia blanca subcortical, que hace que al corte de cerebro se perciba una consistencia anormalmente aumentada para este parénquima.
Respecto a la clínica de los pacientes afectados por panencefalitis diseminada aguda post sarampión, los trastornos de conducta son los síntomas heráldicos del proceso, y es común que como generalmente se presentan en edad escolar, los trastornos del aprendizaje sean la primera manifestación. Comienzan a aparecer posteriormente movimientos mioclónicos generalizados, los cuales tienen traducción EEG, coincidiendo con descargas de alto voltaje. Suele haber también actividad convulsiva.
Por qué, esta paciente de 20 años comenzó con síntomas tanto tiempo después de la infección inicial, y qué elemento desencadenante del trastorno puede haber existido?
Indudablemente, un factor importante puede haber sido, contraer sarampión en etapa muy precoz de la vida (4 meses), momento en que el sistema inmunológico no está plenamente desarrollado, y haber recibido la primera dosis de vacuna 6 meses después. El segundo factor, sin ningún tipo de dudas es el embarazo, condición durante la cual se alteran las condiciones inmunes de la paciente. Respecto de este último punto, digamos que sin pretender analizar la complejidad del sistema inmunológico de la mujer durante el embarazo, el complejo feto/placentario, se comporta como un cuerpo extraño en el útero materno. El feto hereda del padre, antígenos que para el sistema inmune de la madre son extraños. Para no generar una respuesta inmune que pudiera ser dañina para la placenta y el feto, el sistema inmune sufre cambios adaptativos, que le permiten terminar con éxito la gesta. Sin embargo, estos mecanismos adaptativos, a veces pueden comportarse patogénicamente. Los estrógenos y la progesterona, aumentados durante el embarazo, tienen un efecto estimulante sobre la respuesta inmune humoral, pero los corticoides, también aumentados, ejercen un efecto promotor sobre los linfocitos Th2, y suprimen las citoquinas proinflamatorias. En el embarazo existe entonces un aumento de las poblaciones linfocitarias Th2, que explica que se exacerben algunas enfermedades autoinmunes, y que haya mayores manifestaciones alérgicas, y concomitantemente existe una disminución de las poblaciones de linfocitos Th1, lo que trae aparejado que otras enfermedades autoinmunes mejoren (AR, esclerosis múltiple), pero que se produzca a su vez un efecto de disminución en la respuesta inmune celular. Este efecto, podría explicar la reactivación de virus latentes durante años en ámbitos intracelulares, como es el caso del virus del sarampión.
Por último digamos que la panencefalitis esclerosante subaguda es una entidad muy rara hoy en día en el mundo occidental, y sobre todo en los países que han incorporado a su sistema de salud la vacunación antisarampionosa obligatoria. Hay que sospecharla en todo paciente que no registre antecedentes de vacunación, que haya padecido sarampión, y que se comience con sintomatología neurológica caracterizada por comportamiento extraño, deterioro cognitivo, alteraciones emocionales y sociales. Puede haber cambios graduales del comportamiento que se van instalando en meses, que a veces se asocian a espasmos mioclónicos. Puede haber convulsiones, alteraciones de la marcha, y finalmente puede evolucionar al dterioro del nivel de conciencia, coma y muerte.
No existen tratamientos muy efectivos para esta entidad, aunque como se dijo antes, hay intentos con ciertos fármacos que sólo intentan disminuir la progresión de la enfermedad. Hasta ahora, con todos los recursos terapéuticos de los que se dispone, no existen reportes de curas.
Referencias Bibliográficas.
1) al-Eissa A. Sydenham's chorea: a new look at an old disease. Br J Clin Pract 1993;47:14-16. [ISI][Medline]
2) Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002;346:752-763. [Free Full Text]
3) Menge T, Hemmer B, Nessler S, et al. Acute disseminated encephalomyelitis: an update. Arch Neurol 2005;62:1673-1680. [Free Full Text]
4) Thompson EJ, Riches PG, Kohn J. Antibody synthesis within the central nervous system: comparisons of CSF IgG indices and electrophoresis. J Clin Pathol 1983;36:312-315. [Free Full Text]
5) Reiber H. The discrimination between different blood-CSF barrier dysfunctions and inflammatory reactions of the CNS by a recent evaluation graph for the protein profile of cerebrospinal fluid. J Neurol 1980;224:89-99. [CrossRef][ISI][Medline]
6) Reiber H. Flow rate of cerebrospinal fluid (CSF) -- a concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. J Neurol Sci 1994;122:189-203. [CrossRef][ISI][Medline]
7) Reiber H, Peter JB. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci 2001;184:101-122. [CrossRef][ISI][Medline]
8) Case Records of the Massachusetts General Hospital (Case 15-1998). N Engl J Med 1998;338:1448-1456. [Free Full Text]
9) Fishman RA. Cerebrospinal fluid in diseases of the nervous system. 2nd ed. Philadelphia: W.B. Saunders, 1992.
10) Norrby E, Vandvik B. Relationship between measles virus-specific antibody activities and oligoclonal IgG in the central nervous system of patients with subacute sclerosing panencephalitis and multiple sclerosis. Med Microbiol Immunol 1975;162:63-72.
11) Wolinsky JS. Progressive rubella panencephalitis. In: Vinken PJ, Bruyn GW, Klawans HL, McKendall RR, eds. Viral disease. Amsterdam: Elsevier, 1989:405-16.
12) Townsend JJ, Baringer JR, Wolinsky JS, et al. Progressive rubella panencephalitis: late onset after congenital rubella. N Engl J Med 1975;292:990-993. [Abstract]
13) Weil ML, Itabashi H, Cremer NE, Oshiro L, Lennette EH, Carnay L. Chronic progressive panencephalitis due to rubella virus simulating subacute sclerosing panencephalitis. N Engl J Med 1975;292:994-998. [Abstract]
14) Honarmand S, Glaser CA, Chow E, et al. Subacute sclerosing panencephalitis in the differential diagnosis of encephalitis. Neurology 2004;63:1489-1493. [Free Full Text]
15) Swoveland PT, Johnson KP. Subacute sclerosing panencephalitis and other paramyxovirus infections. In: Vinken PJ, Bruyn GW, Klawans HL, McKendall RR, eds. Viral disease. Amsterdam: Elsevier, 1989:417.
16) Gagnon A, Bouchard RW. Fulminating adult-onset subacute sclerosing panencephalitis in a 49-year-old man. Arch Neurol 2003;60:1160-1161. [Free Full Text]
17) Prashanth LK, Taly AB, Ravi V, Sinha S, Arunodaya GR. Adult onset subacute sclerosing panencephalitis: clinical profile of 39 patients from a tertiary care centre. J Neurol Neurosurg Psychiatry 2006;77:630-633. [Free Full Text]
18) Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis 2005;192:1686-1693. [CrossRef][ISI][Medline]
19) Wirguin I, Steiner I, Kidron D, et al. Fulminant subacute sclerosing panencephalitis in association with pregnancy. Arch Neurol 1988;45:1324-1325. [Free Full Text]
20) Gokcil Z, Odabasi Z, Demirkaya S, Eroglu E, Vural O. Alpha-interferon and isoprinosine in adult-onset subacute sclerosing panencephalitis. J Neurol Sci 1999;162:62-64. [CrossRef][ISI][Medline]
21) Anlar B, Yalaz K, Oktem F, Köse G. Long-term follow-up of patients with subacute sclerosing panencephalitis treated with intraventricular alpha-interferon. Neurology 1997;48:526-528. [Free Full Text]
22) Gascon G, Yamani S, Crowell J, et al. Combined oral isoprinosine-intraventricular alpha-interferon therapy for subacute sclerosing panencephalitis. Brain Dev 1993;15:346-355. [CrossRef][ISI][Medline]
23) Yalaz K, Anlar B, Oktem F, et al. Intraventricular interferon and oral inosiplex in the treatment of subacute sclerosing panencephalitis. Neurology 1992;42:488-491. [Free Full Text]
24) Steiner I, Wirguin I, Morag A, Abramsky O. Intraventricular interferon treatment for subacute sclerosing panencephalitis. J Child Neurol 1989;4:20-24. [Free Full Text]
25) Panitch HS, Gomez-Plascencia J, Norris FH, Cantell K, Smith RA. Subacute sclerosing panencephalitis: remission after treatment with intraventricular interferon. Neurology 1986;36:562-566. [Abstract]
26) Ueda S, Okuno Y, Hamamoto Y, Oya H. Subacute sclerosing panencephalitis (SSPE): isolation of a defective variant of measles virus from brain obtained at autopsy. Biken J 1975;18:113-122. [ISI][Medline]
27) Hirano A, Ayata M, Wang AH, Wong TC. Functional analysis of matrix proteins expressed from cloned genes of measles virus variants that cause subacute sclerosing panencephalitis reveals a common defect in nucleocapsid binding. J Virol 1993;67:1848-1853. [Free Full Text]
28) Zaki SR, Bellini WJ. Measles. In: Connor DH, Chandler FW, Schwartz DA, Manz HJ, Lack EE, eds. Pathology of infectious diseases. Stamford, CT: Appleton & Lange, 1997:233-44.
29) Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than previously recognized. J Infect Dis 2005;192:1686-1693. [CrossRef][ISI][Medline]
30) Hummel KB, Lowe L, Bellini WJ, Rota PA. Development of quantitative gene-specific real-time RT-PCR assays for the detection of measles virus in clinical specimens. J Virol Methods 2006;132:166-173. [CrossRef][ISI][Medline]
Diez semanas antes de la internación, la paciente se realizó un test casero de embarazo que fue positivo, consultando a un centro de salud para control de su embarazo. Un test para drepanocitosis, sífilis, HIV, hepatitis B y C fueron negativos. Tests serológicos de IgG para virus varicela-zoster y rubeola fueron positivos. Dos semanas más tarde, una muestra endocervical fue positiva para infección por Chlamydia trachomatis, y negativa para gonorrea. La paciente perdió el turno para consulta y comenzó el tratamiento con azitromicina 4 semanas más tarde.
Seis semanas antes de la internación, se mudó a un asilo para mujeres embarazadas. Los miembros del staff del asilo la describieron como feliz, presentando conducta aniñada, mala memoria, confusión, y movimientos extraños con su cabeza. Durante las siguientes 2 semanas, aparecieron diariamente náuseas y vómitos, que fueron controlados con metoclopramida. Cuatro días antes de la internación,desarrolló mareos y pérdida de fuerzas en el lado izquierdo del cuerpo; comenzó a presentar caídas hacia el lado izquierdo y a presentar vómitos varias veces en el día. Al día siguiente, concurrió a un departamento de emergencias.
En la evaluación en emergencias, la paciente estaba orientada en espacio pero no en tiempo, día ni mes, y brindó datos contradictorios acerca de sus antecedentes médicos. El útero era grávido, y el resto del examen físico era normal. Un electrocardiograma reveló taquicardia sinusal y rotación antihoraria del eje de la onda T en precordiales. Los análisis de orina mostraron proteinuria de 30 mg/dl y un nivel de glucemia de 100 mg/dl. En el screening toxicológico existía positividad para metabolitos tricíclicos en orina.
Una TAC de cerebro reveló una leve prominencia difusa del sistema ventricular. No había masas ni otras lesiones focales.
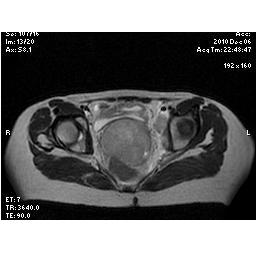
En el segundo día de internación, la paciente estaba alerta, tranquila, cooperativa, y orientada en persona, lugar, y eventos corrientes, aunque no estaba conciente acerca de detalles de su vida. No había temblor, ni signos extrapiramidales. El examen ultrasonográfico reveló una anatomía y crecimiento fetal normales, correspondiente a una gesta de 25 semanas y 6 días. En el tercer día de internación, la debilidad, náuseas y vómitos se resolvieron, y se consideró que había vuelto a su status mental normal. Fue dada de alta al asilo con recomendaciones de seguimiento por consultorio externo de neurología. En el asilo, la paciente volvió a estar mareada, con dificultad para caminar, presentando un episodio de caída de la silla. Esa tarde fue llevada nuevamente al departamento de emergencias.
En el departamento de emergencias, la paciente reportó sentirse “atontada” y nauseosa. Se quejaba de cefalea leve de reciente comienzo, en localización en banda a nivel de la ceja. El interrogatorio mostraba contradicciones; la historia clínica fue provista por los miembros del staff del asilo, pero muchos detalles no pudieron obtenerse. La paciente era nativa de Cabo Verde en el Atlántico Africano, y había migrado a los EE UU hacía 3 años. Había tenido sarampión a los 4 meses de edad y varicela en su niñez. A los 7 años de edad, presentó traumatismo de cráneo en una caída pero dijo haberse recuperado totalmente. Las vacunas, incluyendo la de poliomielitis, difteria, pertusis, y tétanos estaban completas. La vacuna del sarampión la había recibido a los 11 meses; posteriormente, vacunas combinadas de sarampión, parotiditis, y rubeola; y la vacuna contra hepatitis B, 2 años antes de entrar en la escuela secundaria.
Estaba desempleada. Los últimos 3 años había vivido con parientes, amigos, y un novio, así como en asilos. Era soltera y no tenía relación social con el padre de su feto. Sus padres y siete hermanas estaban vivas pero no estaban en contacto con ella. No había historia familiar disponible. No presentaba alergias conocidas, no tomaba alcohol, tabaco ni drogas ilícitas.
En el examen en el departamento de emergencias, la paciente estaba alerta pero no colaboraba, y presentaba movimientos cefálicos involuntarios. Su status mental, según un amigo era el habitual. La presión arterial era de 107/81 mmHg, el pulso de 84 por minuto, y la temperatura de 36,3ºC; la frecuencia respiratoria de 18 por minuto, y la saturación de oxígeno de 100% mientras respiraba aire ambiente. Tenía lesiones acneiformes en la cara. El abdomen era blando, grávido, y no dolía; el feto impresionaba sano. El primer par craneal no fue evaluado, pero del 2º al 12º eran normales. La fuerza era normal, y la marcha insegura. El resto del examen era normal.
 Tabla 1. Resultados de los Tests de Laboratorio.
Tabla 1. Resultados de los Tests de Laboratorio.El segundo día de internación se llevó a cabo una punción lumbar. Los resultados de la punción del líquido cefalorraquídeo se muestran en la Tabla 2; otros resultados se muestran en la Tabla 1. Un ELISA para HIV fue negativo. Un electroencefalograma mostró enlentecimiento difuso de ondas tetha, y una actividad delta rítmica intermitente frontal, que era más prominente en el hemisferio derecho que en el izquierdo. No había actividad epileptiforme (Figura 1). Se repitió una RMN de cerebro después de la administración de gadolinio que no mostró evidencias de realce anormal. Se administró aciclovir por vía intravenosa.

Tabla 2. Resultados de los Tests del Líquido Cefalorraquídeo.

Figura 1. Electroencefalograma Obtenido en el Segundo Día de Hospital.
El EEG es marcadamente anormal, mostrando modesto enlentecimiento del ritmo de base posterior dominante, con salvas de actividad delta rítmica intermitente (flechas), a veces máxima en el lado derecho y otras veces relativamente simétricas bilateralmente. No había elementos epileptiformes ni descargas periódicas.
Al día siguiente, un test sérico para enfermedad de Lyme, un hisopado faríngeo para ácidos nucleicos de Mycoplasma pneumoniae, y cultivos de sangre y orina fueron todos negativos; los resultados de otros tests son listados en la Tabla 1. El quinto día de internación, la condición de la paciente pareció mejorar. Estaba orientada y recordaba detalles de su pasado; la dismetría y la ataxia de tronco se habían reducido. Un nuevo electroencefalograma no mostró cambios. Al día siguiente, se llevó a cabo otra punción lumbar (Tabla 2).
Entre el 7º y el 18º días de hospital, la función motora de la paciente empeoró gradualmente, desarrolló una sensación de que el lado derecho de su cuerpo no le pertenecía, se volvió incapaz de alimentarse por si misma, su capacidad de respuesta y la de seguir órdenes simples disminuyeron, y apareció incontinencia.. Comenzó a adoptar una posición fetal, a emitir gemidos y llantos con gritos ininteligibles. Un test cutáneo para tuberculosis, un hisopado nasofaríngeo en busca de antígenos respiratorios virales y cultivos virales de una muestra de materia fecal fueron todos negativos. Una carga viral para RNA de HIV, así como un test de anticuerpos dirigidos contra HIV fueron negativos. Los niveles de tiroxina total y libre fueron normales, y el nivel de tiroglobulina era elevado, 54,7 ng/ml (normal 4 a 40). En el día 12º, se discontinuó el aciclovir, y se comenzó con ceftriaxona 2 grs EV. Un nuevo estudio electroencefalográfico mostró atenuación aumentada de la actividad de base, y actividad rítmica intermitente frontal delta menos abundante. La RMN en el 13º día, mostró nuevas señales hiperintensas en protuberancia y pedúnculos cerebelosos medios con restricción de difusión de agua asociada en T2 y FLAIR. La restricción de la difusión fue notada en el brazo posterior izquierdo de la cápsula interna izquierda. Había atrofia en el lóbulo temporal medio izquierdo, con resolución de las señales hiperintensas anormales en las imágenes en FLAIR. En el día 14º, una tercera punción lumbar fue llevada a cabo.
Diagnóstico Diferencial:
La paciente vivía semi-independientemente hasta su embarazo, 27 semanas antes de su internación. Su nivel de función cognitiva en ese momento era desconocido, y dado la carencia de información, no fue posible determinar si la afectación de su función eran previas o secundarias a su enfermedad.
El diagnóstico diferencial neurológico se basa primeramente en el examen físico para localizar las lesiones y en el interrogatorio, especialmente la forma de comienzo, y el ritmo de la progresión, para identificar el proceso patológico. El examen neurológico de la paciente mostró una función cognitiva anormal, indicando disfunción de la sustancia gris cortical y subcortical, función motora anormal, indicando disfunción del sistema motor piramidal, y movimientos coreiformes, indicativos de disfunción del sistema motor extrapiramidal. El examen también mostró una marcha torpe, y dificultad en llevar a cabo movimientos alternantes rápidos, indicando disfunción del cerebelo o de sus conecciones. Al no tener información del estado previo de la paciente, no tenemos información del ritmo de instalación de la enfermedad, de su “tempo”, y por lo tanto, necesitamos considerar enfermedades congénitas, y adquiridas que pueden ocasionar evoluciones agudas, subagudas, o crónicas, con episodios de estabilización, o evolución progresiva.
Nosotros hemos basado nuestros diagnósticos diferenciales en el examen neurológico, en los tests iniciales de laboratorio, y en los hallazgos de EEG y RMN.
Examen del Líquido Cefalorraquídeo.
Los resultados del análisis del líquido cefalorraquídeo en esta paciente, mostró una pleocitosis linfocítica, con pocos glóbulos rojos, un nivel levemente elevado de proteínas, y un nivel de glucosa normal. Estos hallazgos son característicos de meningitis aséptica. Por eso nosotros estábamos preocupados por la posibilidad de infecciones por virus, ricketsias, espiroquetas, infecciones decapitadas (parcialmente tratadas), focos parameníngeos de infección, ciertas enfermedades autoinmunes tales como lupus eritematoso sistémico o enfermedad de Behçet, vasculitis, carcinoma, reacciones a los efectos tóxicos de ciertas medicaciones tales como AINES, y meningitis químicas relacionadas con la ruptura de un quiste. Aunque inespecíficos, los hallazgos del líquido cefalorraquídeo proveen sostén a la posibilidad de infección aguda o subaguda, o enfermedad inflamatoria. La presencia de respuesta inflamatoria hace que enfermedades crónicas como enfermedad de Huntington, enfermedad de Wilson, y abiotrofias sistémicas tales como atrofias multisistémicas sean improbables.
Estudios Electroencefalográficos.
El EEG inicial era marcadamente anormal, pero los hallazgos fueron inespecíficos (Figura 1). El ritmo lento dominante y la atenuación posterior sugieren enfermedad de la sustancia gris cortical, mientras que la actividad delta frontal rítmica intermitente sugiere enfermedad de sustancia gris subcortical. Las ondas lentas monomórficas también sugieren que inicialmente la sustancia blanca subcortical estaba relativamente respetada. Dr Henson, puede revisar los estudios radiológicos?
Las imágenes axiales en T2 y FLAIR en la RMN de cerebro el día de la internación, llevadas a cabo sin administración de gadolinio, reveló una región de señal hiperintensa en el lóbulo temporal medio izquierdo (Figura 2A) y un sutil aumento de la señal en el brazo posterior de la cápsula interna izquierda, correspondiente a la localización del tracto corticoespinal. Esos focos no mostraron restricción de la difusión o realce anormal en el estudio llevado a cabo con gadolinio al día siguiente. El aspecto de la protuberancia no era remarcable, y no se notaron otros hallazgos significativos. La venografía por RMN de cerebro fuen normal.

Figura 2. Estudio de Imágenes Cerebrales.
Una RMN en T2 con FLAIR (Panel A), obtenida el día de la internación mostró una región anormal de señal hiperintensa en el hipocampo izquierdo (flecha) y en el brazo posterior de la cápsula interna (no mostrado). El día 13º de internación existía una nueva región de señal hiperintensa en T2 FLAIR en protuberancia y pedúnculos cerebelosos medios (Panel B flechas), y restricción de la difusión en pedúnculos cerebelosos medios, y brazo posterior de la cápsula interna como se muestra en las imágenes de difusión (Panel C, flechas).
Alrededor del día 13º de internación existían marcados cambios. Había una zona de señal anormal en la protuberancia (Figura 2 B), con áreas de difusión restringida. La hiperintensidaddel lóbulo temporal izquierdo medio se había resuelto, y había pérdida de volumen en la región del hipocampo. Había difusión restringida en el tracto corticospinal (Figura 2 C); no se detectaron realces anormales. Estos hallazgos fueron interpretados como resultado de una encefalitis subaguda causada por una infección o un trastorno autoinmune.
En resumen, esta paciente tenía disturbios de la función cognitiva, del haz piramidal, y disfunción cerebelosa, con trastornos del movimiento consistentes en movimientos coreiformes, y tests tanto electroencefalográficos como en las imágenes sugestivos de encefalitis infecciosa o autoinmune.
TRASTORNOS DEL MOVIMIENTO.
La corea es un trastorno hiperkinético del movimiento que puede resultar de un número de enfermedades neurológicas; parece empeorar durante el embarazo, condición conocida como corea gravidarum, o corea gravídica. La mayoría de las pacientes con esta condición, se presentan durante el segundo trimestre del embarazo con movimientos aislados, que se resuelven después del parto. Las causas más comunes son la fiebre reumática aguda (corea de Sydenham), y el síndrome antifosfolipídico. Nosotros también consideramos otras enfermedades asociadas con corea como el lupus eritematoso sistémico, la corea de Huntington, y la enfermedad de Wilson, aunque las últimas dos enfermedades fueron descartadas por los resultados del líquido cefalorraquídeo y por otros elementos.
La corea de Sydenham es una complicación tardía de la infección con estreptococo del grupo A; el comienzo ocurre meses después de la infección aguda. La mayoría de los casos ocurre en la niñez, pero hasta el 30% de los pacientes pueden tener corea recurrente meses o años después del episodio inicial. (1) Esta paciente tuvo sólo un anticuerpo antiestreptolisina mínimamente elevado, sin ninguna otra evidencia de infección o enfermedad cardíaca. El síndrome antifosfolipídico (2) puede ser primario o secundario a lupus eritematoso sistémico, y puede comenzar a manifestarse durante el embarazo. La paciente no tuvo síntomas ni signos de lupus sistémico, pero el lupus confinado al sistema nervioso nervioso central es una entidad perfectamente descripta, y puede empeorar durante el transcurso del embarazo. Los tests para anticuerpos antifosfolípidos fueron todos negativos, así como los tests para lupus.
ENCEFALITIS VIRAL AGUDA.
La encefalitis por herpes simplex fue inicialmente considerada como causa de encefalitis aguda infecciosa en esta paciente dado la anomalía detectada en el hipocampo izquierdo en la RMN. A diferencia de las encefalitis arbovirales y la encefalitis del Oeste del Nilo, que ocurren en el verano y otoño, cuando los mosquitos son abundantes, la encefalitis por herpes simplex ocurre esporádicamente a lo largo de cualquier parte del año. Los resultados negativos de los tests para ácidos nucleicos para virus de herpes simplex en LCR, y la falta de respuesta al aciclovir, hizo que se considerara a este diagnóstico como improbable. Otras causas comunes de encefalitis virales incluyen infecciones por enterovirus y echovirus, así como infecciones por coxscakievirus, que típicamente producen signos prominentes de irritación meníngea, con fotofobia, meningismo, náuseas, y cefalea, pero sólo signos mínimos de disfunción cerebral focal, que pueden ser fugaces y no progresivos. Los tests serológicos en esta paciente, descartaron a estos agentes también como causa.
ENCEFALITIS SUBAGUDAS.
Encefalitis Paraneoplásicas.
La encefalitis límbica paraneoplásica puede preceder a la aparición de un tumor por meses o aún años. Síntomas psiquiátricos, alteraciones en la memoria, confusión y somnolencia, trastornos respiratorios, ataxia, y parálisis de nervios craneales han sido reportados. Hay un modesto aumento de las proteínas en el LCR, pero escasa celularidad. La clínica de esta paciente, así como las alteraciones del LCR no eran consistentes con este diagnóstico.
Encefalitis Postinfecciosa.
Varias infecciones pueden estar asociadas consíndromes de encefalitis postinfecciosa, incluyendo sarampión, parotiditis, y rubeola, influenza, Epstein-Barr, y varicela-zoster. La encefalomielitis diseminada aguda puede ocurrir después de una variedad de infecciones virales y después de la aplicación de la vacuna de la rabia y de la viruela. (3) Usualmente comienza con síntomas inespecíficos tales como fiebre, cefalea, rigidez de nuca, vómitos, y anorexia. El examen neurológico puede mostrar neuritis óptica, ataxia, y signos de foco neurológicos motores; convulsiones y alteraciones de conciencia pueden ocurrir. Esta paciente no tenía antecedentes de inmunizaciones recientes, infecciones virales, o evidencias de neuritis óptica, y las imágenes no eran las típicas vistas en la encefalomielitis diseminada aguda.
El LCR fue una importante pista diagnóstica en este caso. Aunque el nivel inicial de proteínas en LCR estaba levemente aumentado, el componente IgG estaba marcadamente elevado. Dr. Roehrl, podría discutir la importancia de este hallazgo?
El nivel de proteínas totales en LCR, y las IgG, obtenidas el 2º día de internación se muestran en la Tabla 2. La electroforesis en gel de agarosa del LCR reveló bandas oligoclonales en la región gama (Figura 3A). El cociente entre LCR y suero para la albúmina y las concentraciones de IgG fueron QAlb=4.7x10–3 y QIgG=26.1x10–3 , correspondientes a un índice de IgG de 5,6 (valor normal menor de 0,85) (4). En base a este análisis, desarrollado por Reiber, (5,6,7) los resultados de esta paciente (Figura 3B) indicaban marcado aumento de la síntesis intratecal de IgG (fracción de producción intratecal 87,7%) sin evidencias clínicamente significativas de disfunción de la barrera hematoencefálica. La relación de concentración de IgG sobre proteínas totales en LCR fue de 58,9%

Figura 3. Resultados de la Electroforesis del LCR.
El Panel A muestra los resultados de la electroforesis en gel de agarosa de una muestra de LCR de la paciente, obtenida el 2º día de internación (concentrado a 1/63 del volumen original) y un LCR control (concentrado 1/80 del volumen original). P, A, 1, 2, , and denotan regiones electroforéticas de prealbúmina, albúmina, alfa-1, alfa-2, beta, y gama respectivamente. La flecha muestra la posición de varias bandas densas en la región gama, indicando la presencia de múltiples bandas de inmunoglobulinas oligoclonales. Hay una disminución relativa del nivel de albúmina en el LCR de la paciente. El Panel B muestra un gráfico doble logarítmico diseñado de acuerdo al método propuesto por Reiber (también llamado Reibergrama), en el que el cociente entre la concentración en LCR de albúmina y la sérica (QAlb) así como de IgG (QIgG), son graficados en la abscisa y en la ordenada respectivamente. Los límites superiores (QHigh), e inferiores (QLow) de los valores normales son mostrados como líneas sólidas, con la línea de puntos indicando los valores promedio (QMean). Los isopercentiles (líneas discontinuas) corresponden a varias cantidades relativas de producción intratecal de IgG (20 a 80%). La línea discontinua vertical denota el límite superior del QAlb ajustado por la edad, separando la función normal (izquierda) y la función anormal (derecha) de la barrera hematoencefálica. La paciente (punteado rojo) tenía síntesis marcadamente alta de IgG intratecal (fracción de producción intratecal 87,7%) sin evidencias de disfunción significativa de la barrera hematoencefálica (zona 4). La zona 1 denota función normal, la zona 2 denota disfunción pura de la barrera hematoencefálica, la zona 3 denota una combinación de síntesis de IgG intratecal aumentada, y disfunción de la barrera hematoencefálica.
Las enfermedades que pueden provocar una respuesta intratecal de esta magnitud son la sífilis, la panencefalitis rubeólica crónica, y la panencefalitis esclerosante subaguda. (8,9,10,11) Esta paciente no tenía sífilis, como mostraron los tests serológicos y las complejas manifestaciones de esta enfermedad que no son compatibles con sífilis. La encefalitis post-rubeólica (12,13) puede afectar pacientes con antecedentes remotos o infección congénita por rubeola, presentándose como demencia progresiva, ataxia, corea, degeneración retiniana y convulsiones. El examen del LCR muestra pleocitosis, y un nivel de proteínas moderadamente elevado, con hasta un 50% de proteínas del LCR compuestas por inmunoglobulinas. El diagnóstico se confirma por un alto título de anticuerpos anti-rubeola en LCR.
Panencefalitis Esclerosante Subaguda.
El sarampión puede causar tres enfermedades distintas en el sistema nervioso central: encéfalomielitis postinfecciosa, encefalitis subaguda por sarampión, y panencefalitis esclerosante subaguda. (14)
La panencefalitis esclerosante subaguda es típicamente vista entre 7 y 10 años después de una infección por sarampión, y los pacientes se presentan comúnmente con declinación de su rendimiento escolar, cambios conductuales, cefalea, extraños movimientos espontáneos, y a veces convulsiones.(15) Los hallazgos característicos incluyen sacudidas mioclónicas que son a menudo periódicas y están asociadas con descargas epileptiformes uni o bilaterales en el electroencefalograma. Aunque la mayoría de los casos ocurren en la niñez o adolescencia, hay casos que comienzan tan tardíamente como en la quinta década de la vida. (16,17) La incidencia de panencefalitis esclerosante subaguda ha disminuido con la vacunación contra el sarampión; sin embargo, persiste en lugares donde la vacunación no es común. (18) La incidencia de esta condición está aumentada hasta 10 veces, en pacientes que adquieren sarampión antes de la edad de 2 años; esta paciente tuvo sarampión a los 4 meses de edad. La enfermedad puede presentarse durante el embarazo, posiblemente como resultado de la alteración del status inmune. (8,19)
En suma, esta paciente se presentó con una enfermedad neurológica subaguda progresiva, caracterizada por una amplia disfunción del sistema nervioso central, elementos inflamatorios en el LCR, y un extremadamente alto nivel de IgG en el mismo. Nosotros estuvimos de acuerdo en la sala con el diagnóstico de panencefalitis esclerosante subaguda como consecuencia de sarampión adquirido a los 4 meses de edad. Esta condición pudo haber sido exacerbada por el embarazo. Las muestras de suero y de LCR tomadas el 14º día de internación fueron enviadas para testear el nivel de anticuerpos contra sarampión.
El índice de anticuerpos específicos contra sarampión en LCR sobre los del suero estaban elevados a un 31,8 (valor normal menos de 1,4) (7) (Tabla 3). Los anticuerpos IgM específicos contra sarampión, no se detectaron en suero. Este perfil indica una respuesta inmune crónica contra la infeccción sarampionosa en el compartimiento del líquido cefalorraquídeo. Una comparación con los títulos de parotiditis, muestra la especificidad del proceso inmunológico. Los resultados confirman el diagnóstico de panencefalitis esclerosante subaguda en esta paciente. La enfermedad fue una consecuencia tardía de una infección persistente con virus del sarampión.

Tabla 3. Resultados de los Tests para Anticuerpos contra sarampión en Sangre y Líquido Cefalorraquídeo, Obtenidos el Día 14º de Internación.
Discusión del Manejo.
La inmunidad celular estimulando los linfocitos T helper tipo 1 (Th1), inductores de citoquinas es crucial para el clearence del virus del sarampión, la semana después de la infección, mientras que las citoquinas que inducen los linfocitos T helper tipo 2 (Th2) están involucrados en la producción de anticuerpos. Varios reportes han sugerido que el tratamiento con interferón alfa-2 intratecal (una citoquina que promueve la actividad Th1), con o sin tratamiento con el agente inmunomodulador antiviral pranobex, puede disminuir o aún detener la progresión de la panencefalitis esclerosante subaguda. (20,21,22,23,24,25) Sin embargo, no se dispone de datos definitivos que muestren la eficacia de este approach terapéutico.
Cuando hicimos el diagnóstico en esta paciente, ella tenía 28 semanas de gestación, y su condición se estaba deteriorando rápidamente. Nosotros creimos que el tratamiento estaba indicado para mejorar la evolución tanto de la madre como del feto, así que la tratamos con interferón e inosina pranobex por 8 semanas, no obteniendo ningún beneficio claro. Después del parto de un niño sano, por cesárea a las 34 semanas de gestación, se tomó la decisión de discontinuar el tratamiento, con el consentimiento de la madre de la paciente. La paciente falleció 6 semanas más tarde.
Diagnóstico: Panencefalitis Esclerosante Subaguda.
Discusión Anátomo-Patológica.
La autopsia reveló infiltrados inflamatorios conteniendo macrófagos, células plasmáticas, y linfocitos alrededor de los vasos, con destrucción neuronal y gliosis reactiva, más prominentes en tronco cerebral. (Figura 4A) No había inclusiones virales de las típicamente vistas en la encefalitis aguda por sarampión (y ocasionalmente presentes en la panencefalitis esclerosante subaguda) en núcleo o citoplasma tanto en neuronas como en células de la glía, y células endoteliales vasculares. A diferencia de otras enfermedades asociadas al sarampión, la panencefalitis esclerosante subaguda es causada por la persistencia de un tipo defectuoso de virus, que no forman partículas virales completas, pero que pueden infectar células adyacentes por contacto directo. (26,27) En la corteza cerebral, las poblaciones neuronales estaban preservadas, pero había marcada gliosis con astrocitos reactivos (Figura 4B). El tronco cerebral estaba reblandecido por el proceso patológico, pero la sustancia blanca hemisférica era más dura que lo normal; esto era evidente cuando se hacían cortes de cerebro en estado fresco. La dureza correspondía a la eclerosis de la por ese motivo llamada panencefalitis esclerosante subaguda. La sustancia blanca subcortical también mostró gliosis reactiva y extensa activación de la microglia (Figura 4C). Estos hallazgos son inespecíficos, pero junto con los estudios serológicos, son consistentes con el diagnóstico de panencefalitis esclerosante subaguda.

Figura 4. Hallazgos Cerebrales en la Autopsia.
Un marcado infiltrado inflamatorio alrededor de los vasos y destrucción neuronal son los más prominentes en estos cortes del tronco cerebral, que contiene macrófagos , células plasmáticas, y linfocitos; hay prominentes infltrados perivasculares de linfocitos (Panel A, hematoxilina-eosina). En la corteza cerebral, las poblaciones neuronales están preservadas, pero está presente una marcada gliosis (Panel B, análisis inmunohistoquímico para proteínas ácidas fibrilares). Marcada activación de la microglia está presente en la sustancia blanca subcortical con células con formas de varas CD68 positivas (Panel C, flechas; hematoxilina-eosina) (inserto, tinción inmunohistoquímica para CD68).
Las muestras fijadas y congeladas de tejido cerebral fueron enviadas al Dr. William Bellini al Centers for Disease Control and Prevention para estudios inmunohistoquímicos y moleculares. (14,28,29,30) El análisis inmunohistoquímico de antígeno nucleoproteico de virus de sarampión fue negativo. Los estudios diagnósticos moleculares, llevados a cabo para detectar porciones del genoma de virus de sarampión fue negativa en repetidos intentos de amplificación de regiones de genesde nucleoproteínas de sarampión. Débiles señales fueron detectadas en ensayos de PCR de tiempo real, pero fueron insuficientes cantidades de productos de DNA amplificado para llevar a cabo un análisis de la secuencia. Como resultado, no fue posible establecer definitivamente la presencia de virus de sarampión en este caso.
Dos años antes, la paciente había sido vista en un departamento de emergencias en otro hospital por episodios de pérdida de conciencia asociados a movimientos oculares y arqueando el dorso. En el examen presentaba movimientos involuntarios intermitentes de la cabeza y cuello, así como sacudidas en los brazos. Los reflejos osteotendinosos eran vivos y simétricos. Una TAC de cráneo y un EEG fueron normales. Se diagnosticó embarazo temprano en ese momento, y el mismo fue interrumpido por decisión de la paciente. Durante los siguientes 20 meses, la paciente fue perdida en el seguimiento médico, y no se sabe si los movimientos anormales habían continuado.
Es posible que la panencefalitis esclerosante subaguda haya comenzado con ese embarazo previo, se haya estabilizado con la interrupción del mismo, y haya empeorado con el segundo embarazo?
La panencefalitis esclerosante subaguda es casi siempre una enfermedad crónicamente progresiva, aunque están descriptos períodos de plateau en el curso clínico. La duración de la enfermedad puede ser de años, sin embargo, y retrospectivamente, sus síntomas 2 años antes pueden haber sido manifestaciones tempranas de la misma enfermedad.
Diagnóstico Anatómico:
Panencefalitis Esclerosante Subaguda, Secundaria a Infección por Virus del Sarampión.
Fuente
From the Neurology Service (A.J.C., J.W.H., M.P.F.) and the Departments of Radiology (J.W.H.) and Pathology (M.H.A.R., M.P.F.), Massachusetts General Hospital; and the Departments of Neurology (A.J.C., J.W.H.) and Pathology (M.H.A.R., M.P.F.), Harvard Medical School.
Conclusiones del Caso.
Las encefalitis responden a un número muy diverso de causas, que incluyen especialmente agentes infecciosos tales como muchos virus: herpes simplex (HSV 1 y 2), virus de la varicela (VZV), sarampión, parotiditis, rubéola, herpes virus humano tipo 6, algunos agentes bacterianos como micoplasmas, hongos, y parásitos (Toxoplasma gondii) entre otros, y también, causas no infecciosas.
Se trata de una inflamación del parénquima cerebral que puede estar afectado tanto anátomo-patológica como clínicamente en forma focal o difusa. Muchas veces puede acompañarse de signos meníngeos, en cuyo caso se las describe como meningoencefalitis.
Debe diferenciarse desde el punto de vista histológico, de la cerebritis, que es un término utilizado para describir la zona de cerebro en estadio incipiente de formación de un absceso bacteriano.
El curso de las encefalitis puede ser, dependiendo del agente causal y de las condiciones propias del paciente, agudo, subagudo y crónico. Las encefalitis agudas responden más comúnmente a etiologías virales, mientras que las formas subagudas y crónicas más comunes en la actualidad, se ven en pacientes inmunocomprometidos, y probablemente la toxoplasmosis sea el primer factor etiológico en estas dos últimas formas clínicas.
El médico debe estar atento a este tipo de entidades, especialmente al síndrome encefalítico agudo, ya que aunque la mayoría de las veces responden a causas para las que no se dispone hoy en día de tratamientos efectivos, existen dos entidades que son potencialmente tratables. Ellas son la encefalitis por herpes simplex (HSV), que es esporádica y letal tanto en neonatos como en la población sana en general, y la menos frecuente encefalitis por virus de varicela-zoster (VZV), también mortal, que se ve en inmunocomprometidos. Una identificación rápida, asociado a un tratamiento oportuno de estas dos entidades puede salvar la vida del paciente. Es por ello que muchos síndromes encefalíticos agudos son tratados empíricamente con antivirales desde el comienzo de los síntomas, a la espera de la confirmación o no de la causa.
La panencefalitis esclerosante subaguda post sarampión, es una entidad rara en la actualidad, sobre todo desde la implementación de programas de vacunación antisarampionosa obligatoria en la mayoría de los países desarrollados.
El virus del sarampión, que es un RNA virus, perteneciente al grupo de los paramixovirus, tiene un especial trofismo por el sistema nervioso central, y es así que puede afectar al mismo de tres maneras:
• Una forma aguda posinfecciosa, que se produce casi simultáneamente con el exantema, y responde a fenómenos autoinmunes.
• Una forma progresiva aguda, a la que se conoce también como encefalitis por cuerpos de inclusión, que se presenta en pacientes con algún grado de inmunocompromiso y de incapacidad de resolver efectivamente la infección. Se ve tras un corto período de latencia después del exantema.
• Una forma tardía, a veces, como en el caso que nos ocupa, remota, llamada panencefalitis esclerosante subaguda post sarampión. Esta última entidad obedece a mecanismos patofisiológicos no del todo conocidos, pero en los cuales la persistencia del virus o pequeñas secuencias genéticas del mismo (en el caso de la paciente del ateneo no se pudo aislar el virus, ni tampoco antígenos nucleoproteicos, ni una secuencia genómica lo suficientemente específica por técnicas de amplificación de DNA) en el sistema nervioso en forma latente, desencadena una reacción inmunológica dentro del sistema nervioso, caracterizada por un impresionante aumento de las IgG anti virus de sarampión. Este aumento de las IgG específicas en LCR, no tiene una correlación plasmática, lo que indica que se trata de un proceso “barrera hematoencefálica hacia adentro”. Este fenómeno inmunológico local trae como consecuencia una desmielinización preferentemente en localización perivenosa. En el caso específico de la desmielinización en esta entidad, esta se acompaña de una esclerosis (del griego sklerós o “duro”) de la sustancia blanca subcortical, que hace que al corte de cerebro se perciba una consistencia anormalmente aumentada para este parénquima.
Respecto a la clínica de los pacientes afectados por panencefalitis diseminada aguda post sarampión, los trastornos de conducta son los síntomas heráldicos del proceso, y es común que como generalmente se presentan en edad escolar, los trastornos del aprendizaje sean la primera manifestación. Comienzan a aparecer posteriormente movimientos mioclónicos generalizados, los cuales tienen traducción EEG, coincidiendo con descargas de alto voltaje. Suele haber también actividad convulsiva.
Por qué, esta paciente de 20 años comenzó con síntomas tanto tiempo después de la infección inicial, y qué elemento desencadenante del trastorno puede haber existido?
Indudablemente, un factor importante puede haber sido, contraer sarampión en etapa muy precoz de la vida (4 meses), momento en que el sistema inmunológico no está plenamente desarrollado, y haber recibido la primera dosis de vacuna 6 meses después. El segundo factor, sin ningún tipo de dudas es el embarazo, condición durante la cual se alteran las condiciones inmunes de la paciente. Respecto de este último punto, digamos que sin pretender analizar la complejidad del sistema inmunológico de la mujer durante el embarazo, el complejo feto/placentario, se comporta como un cuerpo extraño en el útero materno. El feto hereda del padre, antígenos que para el sistema inmune de la madre son extraños. Para no generar una respuesta inmune que pudiera ser dañina para la placenta y el feto, el sistema inmune sufre cambios adaptativos, que le permiten terminar con éxito la gesta. Sin embargo, estos mecanismos adaptativos, a veces pueden comportarse patogénicamente. Los estrógenos y la progesterona, aumentados durante el embarazo, tienen un efecto estimulante sobre la respuesta inmune humoral, pero los corticoides, también aumentados, ejercen un efecto promotor sobre los linfocitos Th2, y suprimen las citoquinas proinflamatorias. En el embarazo existe entonces un aumento de las poblaciones linfocitarias Th2, que explica que se exacerben algunas enfermedades autoinmunes, y que haya mayores manifestaciones alérgicas, y concomitantemente existe una disminución de las poblaciones de linfocitos Th1, lo que trae aparejado que otras enfermedades autoinmunes mejoren (AR, esclerosis múltiple), pero que se produzca a su vez un efecto de disminución en la respuesta inmune celular. Este efecto, podría explicar la reactivación de virus latentes durante años en ámbitos intracelulares, como es el caso del virus del sarampión.
Por último digamos que la panencefalitis esclerosante subaguda es una entidad muy rara hoy en día en el mundo occidental, y sobre todo en los países que han incorporado a su sistema de salud la vacunación antisarampionosa obligatoria. Hay que sospecharla en todo paciente que no registre antecedentes de vacunación, que haya padecido sarampión, y que se comience con sintomatología neurológica caracterizada por comportamiento extraño, deterioro cognitivo, alteraciones emocionales y sociales. Puede haber cambios graduales del comportamiento que se van instalando en meses, que a veces se asocian a espasmos mioclónicos. Puede haber convulsiones, alteraciones de la marcha, y finalmente puede evolucionar al dterioro del nivel de conciencia, coma y muerte.
No existen tratamientos muy efectivos para esta entidad, aunque como se dijo antes, hay intentos con ciertos fármacos que sólo intentan disminuir la progresión de la enfermedad. Hasta ahora, con todos los recursos terapéuticos de los que se dispone, no existen reportes de curas.
Referencias Bibliográficas.
1) al-Eissa A. Sydenham's chorea: a new look at an old disease. Br J Clin Pract 1993;47:14-16. [ISI][Medline]
2) Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002;346:752-763. [Free Full Text]
3) Menge T, Hemmer B, Nessler S, et al. Acute disseminated encephalomyelitis: an update. Arch Neurol 2005;62:1673-1680. [Free Full Text]
4) Thompson EJ, Riches PG, Kohn J. Antibody synthesis within the central nervous system: comparisons of CSF IgG indices and electrophoresis. J Clin Pathol 1983;36:312-315. [Free Full Text]
5) Reiber H. The discrimination between different blood-CSF barrier dysfunctions and inflammatory reactions of the CNS by a recent evaluation graph for the protein profile of cerebrospinal fluid. J Neurol 1980;224:89-99. [CrossRef][ISI][Medline]
6) Reiber H. Flow rate of cerebrospinal fluid (CSF) -- a concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. J Neurol Sci 1994;122:189-203. [CrossRef][ISI][Medline]
7) Reiber H, Peter JB. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci 2001;184:101-122. [CrossRef][ISI][Medline]
8) Case Records of the Massachusetts General Hospital (Case 15-1998). N Engl J Med 1998;338:1448-1456. [Free Full Text]
9) Fishman RA. Cerebrospinal fluid in diseases of the nervous system. 2nd ed. Philadelphia: W.B. Saunders, 1992.
10) Norrby E, Vandvik B. Relationship between measles virus-specific antibody activities and oligoclonal IgG in the central nervous system of patients with subacute sclerosing panencephalitis and multiple sclerosis. Med Microbiol Immunol 1975;162:63-72.
11) Wolinsky JS. Progressive rubella panencephalitis. In: Vinken PJ, Bruyn GW, Klawans HL, McKendall RR, eds. Viral disease. Amsterdam: Elsevier, 1989:405-16.
12) Townsend JJ, Baringer JR, Wolinsky JS, et al. Progressive rubella panencephalitis: late onset after congenital rubella. N Engl J Med 1975;292:990-993. [Abstract]
13) Weil ML, Itabashi H, Cremer NE, Oshiro L, Lennette EH, Carnay L. Chronic progressive panencephalitis due to rubella virus simulating subacute sclerosing panencephalitis. N Engl J Med 1975;292:994-998. [Abstract]
14) Honarmand S, Glaser CA, Chow E, et al. Subacute sclerosing panencephalitis in the differential diagnosis of encephalitis. Neurology 2004;63:1489-1493. [Free Full Text]
15) Swoveland PT, Johnson KP. Subacute sclerosing panencephalitis and other paramyxovirus infections. In: Vinken PJ, Bruyn GW, Klawans HL, McKendall RR, eds. Viral disease. Amsterdam: Elsevier, 1989:417.
16) Gagnon A, Bouchard RW. Fulminating adult-onset subacute sclerosing panencephalitis in a 49-year-old man. Arch Neurol 2003;60:1160-1161. [Free Full Text]
17) Prashanth LK, Taly AB, Ravi V, Sinha S, Arunodaya GR. Adult onset subacute sclerosing panencephalitis: clinical profile of 39 patients from a tertiary care centre. J Neurol Neurosurg Psychiatry 2006;77:630-633. [Free Full Text]
18) Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis 2005;192:1686-1693. [CrossRef][ISI][Medline]
19) Wirguin I, Steiner I, Kidron D, et al. Fulminant subacute sclerosing panencephalitis in association with pregnancy. Arch Neurol 1988;45:1324-1325. [Free Full Text]
20) Gokcil Z, Odabasi Z, Demirkaya S, Eroglu E, Vural O. Alpha-interferon and isoprinosine in adult-onset subacute sclerosing panencephalitis. J Neurol Sci 1999;162:62-64. [CrossRef][ISI][Medline]
21) Anlar B, Yalaz K, Oktem F, Köse G. Long-term follow-up of patients with subacute sclerosing panencephalitis treated with intraventricular alpha-interferon. Neurology 1997;48:526-528. [Free Full Text]
22) Gascon G, Yamani S, Crowell J, et al. Combined oral isoprinosine-intraventricular alpha-interferon therapy for subacute sclerosing panencephalitis. Brain Dev 1993;15:346-355. [CrossRef][ISI][Medline]
23) Yalaz K, Anlar B, Oktem F, et al. Intraventricular interferon and oral inosiplex in the treatment of subacute sclerosing panencephalitis. Neurology 1992;42:488-491. [Free Full Text]
24) Steiner I, Wirguin I, Morag A, Abramsky O. Intraventricular interferon treatment for subacute sclerosing panencephalitis. J Child Neurol 1989;4:20-24. [Free Full Text]
25) Panitch HS, Gomez-Plascencia J, Norris FH, Cantell K, Smith RA. Subacute sclerosing panencephalitis: remission after treatment with intraventricular interferon. Neurology 1986;36:562-566. [Abstract]
26) Ueda S, Okuno Y, Hamamoto Y, Oya H. Subacute sclerosing panencephalitis (SSPE): isolation of a defective variant of measles virus from brain obtained at autopsy. Biken J 1975;18:113-122. [ISI][Medline]
27) Hirano A, Ayata M, Wang AH, Wong TC. Functional analysis of matrix proteins expressed from cloned genes of measles virus variants that cause subacute sclerosing panencephalitis reveals a common defect in nucleocapsid binding. J Virol 1993;67:1848-1853. [Free Full Text]
28) Zaki SR, Bellini WJ. Measles. In: Connor DH, Chandler FW, Schwartz DA, Manz HJ, Lack EE, eds. Pathology of infectious diseases. Stamford, CT: Appleton & Lange, 1997:233-44.
29) Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than previously recognized. J Infect Dis 2005;192:1686-1693. [CrossRef][ISI][Medline]
30) Hummel KB, Lowe L, Bellini WJ, Rota PA. Development of quantitative gene-specific real-time RT-PCR assays for the detection of measles virus in clinical specimens. J Virol Methods 2006;132:166-173. [CrossRef][ISI][Medline]