INTRODUCCIÓN.
La enfermedad de Behçet (EB), también conocida como
enfermedad de la Ruta de la Seda debido a su ocurrencia en Turquía, Irán,
India, Pakistán, China, Corea y Japón, esto es la antigua ruta comercial de la
seda, en realidad fue ya mencionada por Hipócrates aunque fue descripta por
Hulusi Behçet en 1937.

Figura. Hulusi Behçet y la “ruta de la seda”
Es una enfermedad inflamatoria caracterizada por úlceras
aftosas orales recurrentes y potencialmente numerosas manifestaciones
sistémicas. Ellas incluyen úlceras genitales, lesiones en piel, oculares,
neurológicas, vasculares, gastrointestinales y artritis. Muchas pero no todas
las manifestaciones clínicas de la EB, se consideran debidas a vasculitis. Entre las vasculitis sistémicas, la EB es
particular por su capacidad de comprometer vasos de todos los tamaños
(pequeños, medianos y grandes) tanto en la circulación arterial como venosa.
EPIDEMIOLOGÍA.
La EB es más común (y a menudo más severa) a lo largo de
la llamada antigua “ruta de la seda”, que se extiende desde el este de Asia
hasta el Mediterráneo (1) Es más común en Turquía (80 a 370 casos por 100.000),
mientras que la prevalencia va de 13,5 a 20 por 100.000 en Japón Corea, China,
Irán, y Arabia Saudita (1). La prevalencia en París, Francia en 2003 fue 7,1
por 100.000. Es algo más común en hombres quye en mujeres, y típicamente afecta
a varones de 20 a
40 años de edad (1).
ETIOLOGÍA Y PATOGENIA.
La causa de la EB es desconocida. Como con otras
enfermedades autoinmunes, el trastorno puede representar actividad inmune
aberrante disparada por la exposición a un agente, quizás infeccioso, en
pacientes con predisposición genética para desarrollar la enfermedad. Los
mecanismos mayores de enfermedad en la EB incluyen los siguientes (5):
·
Influencias genéticas incluyendo
asociación con algunos antígenos de histocompatibilidad (HLA).
·
Antígenos bacterianos con reactividad
cruzada a péptidos humanos.
·
Poblaciones alteradas de células
hemopoyéticas y citoquinas asociadas.
·
Presencia de inmunocomplejos y
autoanticuerpos.
·
Activación vascular endotelial e
hipercoagulabilidad.
Los agentes disparadores propuestos incluyen antígenos
virales y bacterianos, o de origen ambiental, tales como químicos o metales
pesados. Aunque es todavía un contribuyente posible y no se descarta como
mecanismo, la infección directa de los tejidos afectados por algún agente no se
ha observado.
La predisposición genética es probablemente poligénica.
Hay asociación entre la EB y el HLA-B51/B5 (6).
Aunque la mayoría de los casos de EB son esporádicos, se
han reportado casos en familias con
múltiples miembros afectados. Hay otros genes no-HLA que también juegan algún rol en la susceptibilidad a la
enfermedad. Ellos son: un polimorfismo en el gen de las moléculas de adhesión
intercelular (ICAM)-1, el gen de la óxido nítrico sintasa, los genes del factor
de necrosis tumoral (TNF), los genes del factor de crecimiento endotelio
vascular (VEGF), de la superóxido dismutasa, del citocromo P450, y muchos otros
genes (10,18,34-53). Mutaciones sin sentido del gen de la fiebre Mediterránea
familiar que codifica para la proteína pirina es expresado en la superficie de
los neutrófilos (54-57).
El mimetismo molecular entre ciertos antígenos bacterianos
que tienen reactividad cruzada con péptidos humanos, parecería tener también
algún rol patogénico. Los autoantígenos incluyen las proteínas del shock
térmico, una familia de proteínas de 60 a 90 kDa producidas por muchas células en
respuesta al stress celular (58,59). Una
respuesta exagerada a las proteínas del shock térmico de micobacterias por
parte de las células T gama-delta puede jugar un rol en le EB (60). Bacterias
específicas, particularmente estreptococos pueden ser importantes
contribuyentes a la respuesta cruzada en la EB (61,62,63,64,65,66,67,68,69).
También se ha implicado a Helicobacter pylori (70), al virus del herpes simplex
(HSV), y al parvovirus B19.
Hay en le EB alteraciones de la función inmune innata. La
lectina de unión a la manosa (MBL) es parte del sistema inmune innato que
activa la cascada del complemento después de unirse a carbohidratos de la
estructura de los microorganismos. En la EB de ha visto bajos niveles de MBL y
mutaciones del gen de la MBL, como también se ha visto en otras enfermedades autoinmunes
como el lupus eritematoso sistémico y la artritis reumatoidea. Muy bajos
niveles se correlacionan con mayor severidad de la EB. Dado que niveles bajos
se relacionan con mayor susceptibilidad a las infecciones, y dado que la EB se
asocia a algunas infecciones, este podría ser el nexo explicativo de tal
relación (62,71).
Los receptores toll-like son receptores de los linfocitos
y de algunos tejidos sólidos dirigidos contra microbios patogénicos y son parte
del sistema inmune. En la EB se ha visto patrones alterados de los receptores
toll-like (72,73,74)
También se han visto alteraciones en la inmunidad celular
y en las citoquinas en la EB. Existen en la EB alteraciones en el número de
subpoblaciones de células T y evidencias de activación celular (5). Las células
T autorreactivas parecen jugar un rol crítico en la patogénesis de la EB
(75,76-78).
Una respuesta T helper 1 predominante se ha observado,
aunque también se han visto evidencias de activación de respuesta TH2. Por lo
tanto, habría un mix de reactividad TH1 y TH2 en la EB (13,79-81).
Los linfocitos TH1 que producen las citoquinas: IL-2,
IL-6, IL-8, IL-12, IL-18, TNF alfa, e interferon gama están aumentados en
pacientes con EB (13,79,82-98).
Los cambios asociados al fenotipo TH2 han sido también
observados. Se ha visto en algunos pacientes, que células mononucleares de la
sangre periférica producen cantidades aumentadas de IL-4, IL-10, e IL-13
(29,80,99-119).
Los autoanticuerpos y la formación de inmunocomplejos en
la EB son evidencias de la participación de la inmunidad humoral en la EB. Se
ha visto en EB un aumento de los linfocitos B circulantes que parecerían
dirigidos a una respuesta antigénica específica y no simplemente una respuesta
policlonal (13,120,121).
Los autoanticuerpos parecen jugar un rol en la EB. Estos
están dirigidos contra un número de tejidos (targets), incluyendo antígenos de
la mucosa oral, células endoteliales, moléculas co-estimulatorias de céluas T
como CTLA-4, receptores killer inmunoglobulina-like, LDL oxidadas, y kinectina
(122-127). Anticuerpos anti- saccharomyces cerevisiae (ASCA) se han visto en EB
con la misma frecuencioa que en la
espondilitis anquilosante y en la colitis ulcerosa, pero no tanto como en la
enfermedad de Crohn (122-128).
La actividad autoinmune contra autoantígenos retinianos
parece ser importante en la patogénesis de la uveítis de la EB
(129,130,131,132-135).
Otro tópico importante relacionado con la EB es la
activación endotelial y las alteraciones de la coagulación. La disfunción
endotelial es característica de la EB (138-142), y es causa de inflamación
vascular así como de trombosis. Hay un número de moléculas identificadas, que
son responsables de la disfunción endotelial en la EB. El óxido nítrico (NO) es
una molécula altamente reactiva asociada con la actividad inflamatoria y la
función endotelial. En la EB se ha visto niveles aumentados de NO en suero,
eritrocitos, sinovial, y humor acuoso (123,143-148). Se ha visto también en EB
de un stress oxidativo aumentado (139,149).
Se han visto en EB aumentos de los niveles de homocisteína
plasmática (150,151). La hiperhomcistinemia puede ser adquirida y ser
potencialmente reversible. Se asocia a trombosis y también se la considera un
marcador de actividad de la EB (13,118,152-154).
Los niveles de factor de crecimiento endotelio vascular (VEGF),
son mayores en pacientes con EB, y se han asociado con aumentos de actividad de
la enfermedad y con las concentraciones de NO (51,155-157). Los niveles de VEGF
son mayores en el líquido cefalorraquídeo de pacientes con EB y compromiso
neurológico que en pacientes con EB sin el mismo (158).
Se han encontrado anticuerpos IgM anti-endotelio (160).
Esto genera activación endotelial con un estado hipercoagulable con aumento de
los niveles de factorr VIII, factor XI, factor de von Willebrand y ristocetina,
antitrombina III, y fibrinógeno (163). La fibrinolisis está disminuida
(13,164,165,166)). Hay menores concentraciones de proteína C y de
trombomodulina soluble en el plasma de pacientes con EB. La trombomodulina,
presente en la superficie de las células endoteliales, se une a la trombina,
cambiando así la especificidad del sustrato natural de la trombina (el
fibrinógeno). De esta unión (trombina-trombomodulina) se produce la activación
de la proteína C (que es un potente anticoagulante), en vez de clivar el
fibrinógeno.
La actividad plaquetaria está aumentada en pacientes con
EB (167), y también se ha visto disminución de la deformabilidad eritrocitaria
(168,169)Los niveles de activador tisular del plasminógeno son emnores en
pacientes con EB y trombosis venosa profunda, lo que sugiere un defecto en la
fibrinolisis (170-173).
La mayoría de las evidencias actualmente disponibles
sugieren que la patogénesis de la EB es probablemente no debida a estado
hipercoagulable sino más bien a daño vascular inducido por inflamación, o
disfunción endotelial intrínseca que puede servir como estímulo trombogénico en
si mismo (174-176).
Los pacientes con EB tienen un aumento del grosor
íntima-media, y disminución de la distensibilidad arterial comparado con los
controles (177).
Por último, se ha visto activación de los
polimorfonucleares (PMN) neutrófilos en la EB. La motilidad de los PMN está
aumentada en EB debido a aumento de los niveles circulantes de IL-8, y
TNF-alfa, los cuales tamnién activan la superficie endotelial
(178,179,180,181). También hay aumento de la expresón de E-selectina lo que
hace que los neutrófilos se adhieran al endotelio, y facilita la migración en
la pared del vaso afectado y más allá del mismo (141). Hay aumento del ICAM-1
soluble en EB, y su concentración se correlaciona con la actividad de la
enfermedad (71,182,189). Los niveles de los factores estimulantes de colonias
(G-CSF) (190).
Los indicadores de la activación neutrofílica han sido
indicados como posibles marcadores de actividad y severidad de la enfermedad.
Ellos incluyen la elastasa de polimorfonucleares, la mieloperoxidasa
plasmática, y los productos de oxidación proteica avanzada
(77,191,192,194,195,196).
ANATOMÍA PATOLÓGICA.
El examen histológico de tejidos afectados por EB revela
frecuentemente vasculitis aunque este hallazgo puede no ser demostrado en todas
las lesiones (5,197). Las clásicas lesiones de la EB son la perivasculitis
obliterativa leucocitoclástica necrotizante y la trombosis venosa con
infiltrado linfocitario de los capilares en venas y arterias de todos los
tamaños. El infiltrado celular es a menudo de distribución perivascular; los
neutrófilos y los linfocitos T CD4+ están presentes alrededor de los vasa
vasorum y área perivascular. Cuando hay una franca vasculitis leucocitoclástica
puede haber inflamación endotelial, extravasación de eritrocitos, y necrosis
fibrinoide de la pared de los vasos. La
trombosis está a menudo presente (13).
Las lesiones mucocutáneas demuestran infiltración
linfocitaria, inmunoglobulinas y deposición del complemento, con
licuefacción-degeneración en la unión dermo-epidérmica asociada a la formación
de úlceras (198,199).
El infiltrado neutrofílico y mononuclear es observado
frecuentemente en lesiones agudas espontáneas y siguiendo a un test de patergia
(5,200). Las lesiones pápulo-pustulares pueden mostrar vasculitis
leucocitoclástica con IgM, IgG, C3, y depósitos de fibrina consistentes con
vasculitis por complejos inmunes (201,202). Las lesiones pustulares de piel son
a menudo no estériles y pueden contener Staphylococcus aureus especies de
Prevotella (203).
La lesión tipo eritema nodoso en la EB demuestra paniculitis
septal, lobulillar, o mixta, lobulillar y septal, un número variable de neutrófilos,
linfocitos e histiocitos, y a menudo muestra vasculitis leucocitoclástica.
Las lesiones oculares de la EB pueden mostrar infiltrado
leucocitario alrededor de los vasos, perivasculitis oclusiva retiniana, y
trombosis. La panuveítis no granulomatosa es característica. Durante la
actividad de la enfermedad, los neutrófilos pueden estar presentes en la cámara
anterior del ojo así como en el epitelio corneano, iris, cuerpos ciliares y
coroides (13).
Las lesiones en el sistema nervioso central en la EB demuestran
infiltrados de linfocitos T, monocitos y apoptosis de las neuronas afectadas
(204). Las áreas comprometidas incluyen la médula espinal, el tronco cerebral
(incluyendo protuberancia, bulbo y mesencéfalo), cerebelo, ganglios de la base,
tálamo, cápsula interna, y sustancia blanca periventricular.
Aunque el compromiso renal es infrecuente y usualmente
leve, cuando está presente puede mostrar proliferación mesangial,
glomerulonefritis proliferativa y formación de semilunas; depósitos de
inmunoglobulinas, complemento, y complejos inmunes en el glomérulo (205).
La artritis de la EB se caracteriza por inflamación
sinovial con mayor infiltrado neutrofílico que linfocitario (194).
MANIFESTACIONES CLÍNICAS.
El hallazgo común en pacientes con EB es la presencia de
úlceras mucocutáneas dolorosas recurrentes. Otras manifestaciones varían de
acuerdo a las poblaciones.
La severidad es mayor en hombres. La mayor morbimortalidad
ocurre cuando existe enfermedad ocular (que afecta a 2/3 de los pacientes), enfermedad
vascular (que afecta 1/3 de los pacientes), y sistema nerviso central (que
afecta 10 a
20 por ciento de los pacientes). Las manifestaciones cutáneas y articulares son
comunes. La enfermedad renal y del sistema nervioso periférico son raros
comparados con las vasculitis (216).
Aunque hay datos limitados en niños con EB, las
manifestaciones clínicas parecen ser similares a los adultos (208,217).
Úlceras orales.
La mayoría pero no todos los pacientes se presentan
inicialmente con úlceras aftosas recurrentes, que son macroscópicamente e
histológicamente similares a las úlceras aftosas recurrentes de la estomatitis
aftosa recurrente pero que tienen tendencia a ser más grandes, y a menudo
múltiples (Figura 1, y Figura 2).

Figura 1. Múltiples aftas orales dolorosas en un paciente
con enfermedad de Behçet.
Las úlceras son característicamente dolorosas, y en casos
severos pueden comprometer la alimentación. Son redondeadas y van desde pocos
milímetros hasta 2
centímetros . Las úlceras menores son definidas como de
menos de 1 cm ,
y las úlceras mayores como mayores a 1 cm . La porción
externa de los labios no está comprometida.

Figura 2. Múltiples úlceras orales en un paciente con
enfermedad de Behçet.
Se requiere que las úlceras orales recurran más de 3 veces
en un año para poder ser utilizadas como criterio de EB. La curación de las
úlceras es típicamente espontánea dentro de una a tres semanas; cuando las
lesiones son recurrentes sin embargo, algunos pacientes tienen úlceras en forma
continua. Las úlceras orales son típicamente la primera manifestación de la
enfermedad de Behçet, y se presentan en forma intermitente durante muchos años
para hacerse menos comunes después de 20 años de enfermedad (211). Los
fumadores con EB pueden tener menos incidencia de úlceras (218).
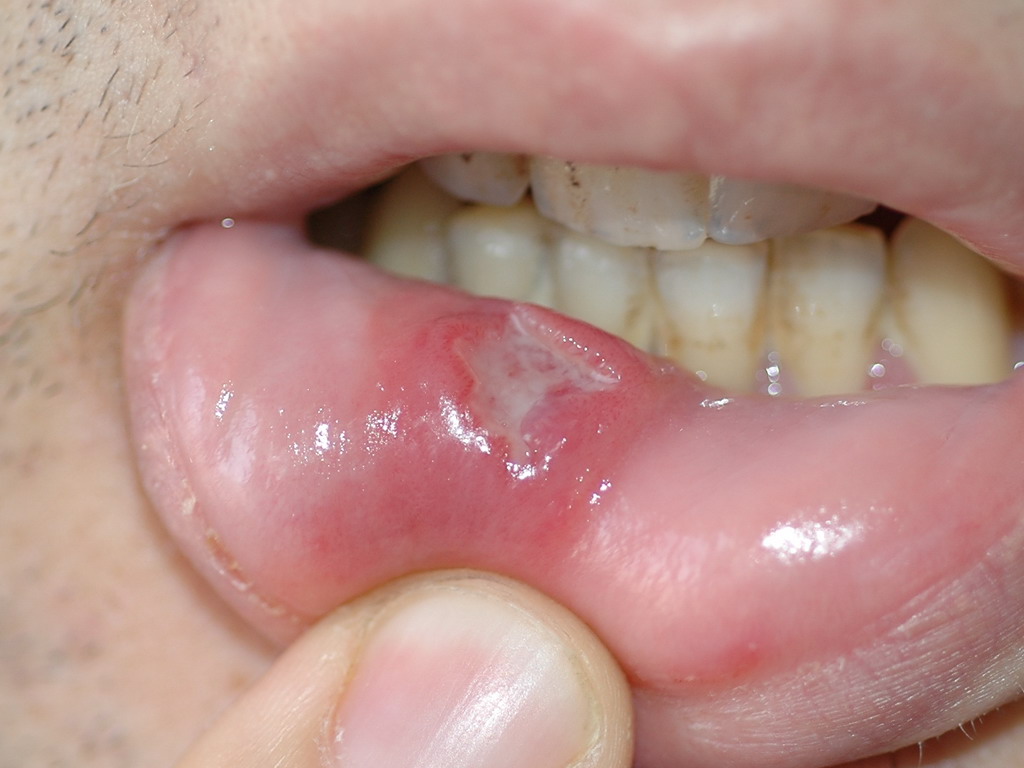
Figura 3. Úlcera en labio inferior de gran tamaño en un
paciente con enfermedad de Behçet.
Lesiones urogenitales.
Las ulceraciones genitales ocurren en 75 por ciento o más
de pacientes con EB. Las úlceras son similares en apariencia a las aftas orales
y usualmente son dolorosas. Se encuentran más comúnmente en el escroto en el
hombre y en la vulva en la mujer (Figura 4, 5, y 6). La recurrencia es
típicamentemenos frecuente que las úlceras orales. La formación de cicatrices
es frecuente para las lesiones genitales. Las cicatrices en escroto, secundaria
a la curación de una úlcera, raramente si es que puede ser, se ven en otra
condición distinta que la EB.

Figura 4. úlceras vasculíticas en enfermedad de Behçet.
Úlceras vasculíticas dolorosas de escroto, pene y dedos de
manos en un paciente con enfermedad de Behçet.
Figura 5. Enfermedad de Behçet con extensa úldera
escrotal.

Fig 6. Extensa úlcera escrotal en paciente con enfermedad
de Behçet.
Lesiones cutáneas.
Las lesiones cutáneas también ocurren en un 75 por ciento
de los pacientes con enfermedad de Behçet. Las manifestaciones de piel varían y
pueden incluir lesiones acneiformes (Fig 7, 8), erupciones
pápulo-vesículo-pustulares, pseudofoliculitis, nódulos, eritma nodoso
(paniculitis septal) (Fig 9), tromboflebitis superficial, lesiones tipo
pioderma gangrenoso, lesiones tipo eritema multiforme, y púrpura palpable. La
biopsia de las lesiones de eritema nodoso revela paniculitis septal con
vasculitis de vasos de mediano calibre en hasta la mitad de las lesiones (221).

Figura 7. Lesión acneiforme en un paciente con enfermedad
de Behçet.
Las lesiones acneiformes son más comunes en aquellos
pacientes que tienen artritis (222,223).

Figura 8. Lesiones acneiformes en enfermedad de Behçet.

Figura 9. Eritema nodoso en un paciente con enfermedad de
Behçet.
Esas lesiones consisten en pápulas y pústulas
indistinguibles del acné ordinario, y comparte características de flora
microbiológica con las lesiones pápulopustulares del acné (224). Las lesiones
pustulares de piel son a menudo no estériles y pueden contener Staphylococcus
aureus y Prevotella spp (224).
La patergia se refiere a lesiones eritematopapulares o
pustulares en respuesta a injuria de la piel. Se define como una lesión tipo
pustular o una pápula que aparece 48 horas después del pinchazo de una zona de
piel con una aguja estéril de calibre 20 a 21 (Figura 10,11,12). La patergia es menos
común en Estados Unidos o Europa (10
a 20 por ciento) que en áreas más endémicas (50 a 75 por ciento). El
dermografismo es una respuesta al raspado suave de la piel que puede estar
presente en algunos pacientes (225) (Fig 13).

Figura 10. Test de patergia.
1) momento en el que el paciente es pinchado con una aguja estéril; 2) muestra
el área inmediatamente después del pinchazo; (3 y 4) muestran el área un día y
dós días después del pinchazo respectivamente, en un paciente con test de
patergia positivo.

Figura 11. Otro caso de test de patergia positivo.

Figura 12. Técnica y profundidad del pinchazo en el test
de patergia.

Figura 13. Dermografismo
positivo.
Las alteraciones capilares en el lecho ungueal ocurren en
75 por ciento de los pacientes (226). El grado de anormalidad se correlaciona
con el grado de enfermedad de piel y artritis.
La enfermedad ocular ocurre en 25 a 75 por ciento de los
pacientes con EB, dependiendo de la población estudiada, y en la mayoría de los
casos puede progresar a la ceguera si no se trata. La enfermedad es típicamente
en países no endémicos de EB con menor incidencia de pérdida de visión (227).
La uveítis es a menudo el hallazgo dominante. Es
típicamente bilateral y episódica. A menudo compromete todo el tracto uveal
(panuveítis), y puede no resolverse completamente entre los episodios.
El hipopion es una severa uveítis anterior (Figura 14 A y B) con material
purulento en la cámara anterior del ojo que se ve característicamente en 20 por
ciento de los pacientes con EB. Muchos pacientes con hipopion presentan
vasculitis retiniana (228).

Fifura 14 A . Hipopion
(flecha). Una línea horizontal de células inflamatorias en la cámara anterior
del ojo y deformidad del iris. El círculo brillante es la luz reflejada.

Figura 14 B. Hipopion. Un niño con uveítis anterior e
hipopion bilateral (una capa de pus en la cámara anterior del ojo). Aunque las
uveítis anteriores como esta pueden llevar a la ceguera, esta complicación es
mucho más común en las uveítis posteriores, que afectan el cuerpo vítreo y la
retina.
La uveítis posterior, la vasculitis retiniana (Figura 15 A y B), oclusión vascular y
la neuritis óptica requieren tratamiento inmunosupresor y pueden afectar
irreversiblemente la visión y progresar a la ceguera si no son tratados.

Figura 15
A . Uveítis en enfermedad de Behçet. Amplia hemorragia
retiniana de un paciente con Behçet. Las hemorragias son secundarias a
vasculitis retinianas.

Figura 15 B. Isquemia retiniana en enfermedad de Behçet.
La imagen muestra isquemia retiniana avanzada y palidez del nervio óptico en un
paciente con EB y varios ataques previos de severa enfermedad vasooclusiva.
Otros cambios que pueden ser vistos incluyen
neovascularización, cataratas secundarias, glaucoma, y en aproximadamente 3 por
ciento de los pacientes, ulceración conjuntival (229, 230). La
neovascularización, ocurre generalmente
debido a inflamación, y el tratamiento con inmunosupresores, particularmente
con interferón puede ser beneficioso (231). La conjuntivitis, escleritis,
epiescleritis, y el síndrome de sicca son raros.
Enfermedad neurológica.
La enfermedad neurológica ocurre en menos de un quinto de
los pacientes con EB (233-236) Se observa más frecuentemente en hombres que en
mujeres. Las lesiones parenquimatosas focales y las complicaciones
trombóticas vasculares son las alteraciones más frecuentes. Otras
manifestaciones incluyen las meningitis asépticas o las encefalitis y las
vasculitis arteriales. Los cambios de personalidad, alteraciones psiquiátricas
y la demencia pueden hacerse presentes. A diferencia de otras vasculitis
sistémicas, la neuropatía periférica no es común en la EB, aunque puede
desarrollar en un subgrupo de pacientes (237-239).
Las manifestaciones neurológicas focales pueden ser
debidas a lesiones en el haz piramidal, en el tronco cerebral, en la sustancia
blanca periventricular, en la médula espinal y en los ganglios de la base. El
compromiso cerebeloso, aunque está descripto, es muy infrecuente. Esas lesiones
del sistema nervioso central son mejor detectadas por RMN (233,240). La
anatomía patológica demuestra infiltrado linfocitario perivenular, infiltración
por células inflamatorias, gliosis, necrosis, y pérdida neuronal. Aunque no
siempre se observa vasculitis franca en las lesiones parenquimatosas, a veces
se ven en vasos cerebrales más grandes, incluyendo arterias o venas (Figura 16,
y Fig 17). Las arteritis pueden producir strokes isquémicos, dilataciones
aneurismáticas, y hemorragia subaracnoidea.

Figura 16. Neuro-Behçet. Compromiso
parenquimatoso cerebral. RMN
En T2 (A) y en coeficiente de difusión aparente (B)
obtenida tres días después del inicio se observa hiperintensidad y difusión aumentada
que afecta los ganglios de la base del lado derecho. Las imágenes de
seguimiento (Cy D), obtenidas 1 año más tarde muestra considerable resolución
de las alteraciones previas en T2 y difusión, pero muestran atrofia de los
ganglios basales. 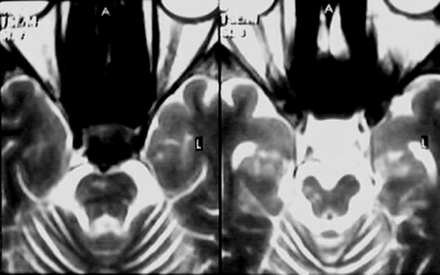
Figura 17. Neuro-Behçet.
Compromiso
parenquimatoso cerebral.
RMN en T2 mostrando múltiples placas hiperintensas en
protuberancia y en el lado derecho del tronco cerebral en un paciente con
enfermedad de Behçet.

Figura 18. Venografía por resonancia magnética que muestra
obliteración de la porción media del seno sagital superior en un paciente con
enfermedad de Behçet que se presentó con hipertensión intracraneana.
En una gran serie de casos de 200 pacientes con EB donde
se reportó compromiso neurológico (233), se vio que en promedio, el compromiso
neurológico aparecía entre cinco a seis años de la primera manifestación de
enfermedad. No obstante, un 7,5 por ciento pueden comenzar simultáneamente con
el comienzo de la enfermedad, o incluso (3 por ciento), preceder a las
manifestaciones extraneurológicas de la enfermedad. Veinte por ciento de los
pacientes con compromiso neurológico estaban asintomáticos.
El pronóstico varía según el tipo de proceso neurológico.
Aquellos con trombosis de los senos durales u otros procesos no
parenquimatosos, menos probablemente tenga enfermedad recurrente, discapacidad
o muerte prematura. En comparación, los pacientes con compromiso parenquimatoso
tienen peor pronóstico (233,244).
El líquido cefalorraquídeo puede ayudar a determinar el
pronóstico. En un estudio, 90 por ciento de aquellos pacientes con un nivel
elevado de proteínas o aumento de celularidad en líquido cefalorraquídeo,
tuvieron eventos neurológicos adicionales, discapacidad progresiva, y muerte
durante los tres años de seguimiento.
Mientras que sólo el 25 a
30 por ciento de aquellos con niveles de proteínas normales en LCR sufrieron
alguno de tales eventos (233).
Enfermedad vascular.
La mayoría de las manifestaciones clínicas de la EB se
consideran debidas a vasculitis, y la enfermedad de Behçet se caracteriza por
su tendencia a afectar los vasos de todos los tamaños (pequeños, medianos o
grandes), tanto en el sector venoso como arterial de la circulación.
La enfermedad arterial afecta es más comúnmente una
vasculitis de pequeños vasos, pero puede
producir también vasculitis de mediano o de gran calibre.
El compromiso de los vasos de gran calibre ocurre
aproximadamente en un tercio de los pacientes con EB (245). En esos pacientes,
la inflamación perivascular, y endovascular pueden conducir a hemorragia,
estenosis, formación de aneurismas, y formación de trombos tanto en arterias
como en venas, y várices. La progresión y recurrencia son más probables en
estos pacientes, y el tratamiento inmunosupresor de esta inflamación ha sido
beneficioso aunque hay pacientes que requieren intervención vascular (246).
Enfermedad Vascular en 728 Pacientes con Enfermedad de
Behçet.
Enfermedad Venosa
·
Trombosis venosa profunda (221
pacientes).
·
Tromboflebits subcutánea (205 pacientes).
·
Síndrome
de vena cava superior (122 pacientes).
·
Síndrome
de vena cava inferior (93 pacientes).
·
Trombosis
de senos venosos cerebrales (30 pacientes).
·
Síndrome
de Budd-Chiari (17 pacientes).
·
Otras oclusiones venosas (*) (24
pacientes).
Enfermedad Arterial.
·
Oclusión de arteria pulmonar o aneurisma
de arteria pulmonar (36 pacientes).
·
Aneurismas de aorta (17 pacientes).
·
Oclusión arterial en un miembro o
aneurisma (45 pacientes).
·
Otra oclusión arterial o aneurisma (**)
(42 pacientes).
·
Trombo en ventrículo derecho (2
pacientes).
(*) Otras venas incluyen: subclavias, ilíacas, porta,
renales, innominada, tronco braquiocefálico.
(**) Otras arterias incluyen: ilíacas, subclavias,
renales, carótidas, cerebrales, coronarias, innominadas, mesentéricas, aorta,
basilar, esplénica.
El compromiso arterial pulmonar es particularmente característico, y es
importante el reconocimiento temprano. La hemoptisis es el íntoma de
presentación más común (247). Los aneurismas de la arteria pulmonar afectan las
grandes ramas proximales de las arterias pulmonares, y son las lesiones
vasculares pulmonares más frecuentes en la EB, siendo además raros de ver en
otras enfermedades que no sean EB (Fig 18/1, Fig 19).

Figura 18/1. Aneurisma de la arteria pulmonar en paciente con enfermedad
ede Behçet. En la Rx de tórax se
demuestra la presencia de una masa mal definida de densidad de partes blandas y
contorno regular en la región derecha del hilio pulmonar (A), cuyas dimensiones
se redujeron significativamente después de tratamiento con corticosteroides e
inmunosupresores (B).

Figura 19. Tomografía computada de tórax del mismo paciente de la imagen
superior, después de la administración de contraste iodado, que demostró
dilatación aneurismática de 50
mm en su mayor diámetro axial, con engrosamiento
parietal difuso. Se pueden ver otros aneurismas más pequeños y tamaños
variables, de carácterísticas morfológicas similares, bilaterales,
predominantemente en las regiones centrales y en el tercio superior y medio de
los pulmones.

Figura 20. Síndrome de Hughes-Stovin en un paciente de 35 años con
enfermedad de Behçet. Reconstrucción tridimensional de una angiotomografía
multi-slice que muestra estrechamientos irregulares (flechas) de las arterias
pulmonares, así como dilataciones aneurismáticas (cabezas de flecha) de las
arterias pulmonares periféricas.
El síndrome de Hughes-Stovin puede ser considerado parte del espectro
de la EB, o puede ser considerado un raro trastorno de etiología desconocida
cuando no reúne criterios de EB. También a veces es considerado como
“enfermedad de Behçet frustra”. Ocurre predominantemente en adultos jóvenes
entre la segunda y cuarta décadas de la vida. Los aneurismas de la arteria
pulmonar han sido atribuidos a debilitamiento de la pared de los vasos debida a
inflamación. Los hallazgos asociados incluyen trombos en el sistema de la vena
cava, senos cerebrales, venas de los miembros; oclusiones de las arterias
pulmonares por embolias o trombosis; y uno o más aneurismas de las arterias
pulmonares, asociados frecuentemente a aneurismas de las arterias bronquiales.
Las manifestaciones típicas son vistas en
tres fases: la primera con síntomas de tromboflebitis, un segundo estdio
con formación y agrandamientos de aneurismas pulmonares, y un tercer estadio
con ruptura de los aneurismas que produce hemoptisis masiva y muerte. Hay que
hacer diagnóstico diferencial con otras causas de aneurismas de la arteria
pulmonar o de las arterias bronquiales como aplasia de la arteria pulmonar,
trauma, infección, silicosis y vasculitis (sobre todo enfermedad de Behçet)
La enfermedad venosa en la EB es más común que la arterial. La
oclusión de la vena cava superior e inferior, el síndrome de Budd-Chiari, las
trombosis de los senos durales, y otras lesiones venosas obstructivas pueden
verse, además de las tromboflebitis superficiales y las trombosis venosas
profundas de los miembros que son mucho más frecuentes. Las trombosis venosas
son a menudo una manifestación temprana de la EB. Las trombosis recurrentes de
las extremidades inferiores pueden conducir a síndrome posflebítico (228).
Una respuesta patergia-like puede ser evidente después de procedimientos
vasculares los que resultan en flebitis o aneurismas (253,254).
Artritis.
Una artritis no deformante, no erosiva, asimétrica ocurre usualmente
en la mitad de los pacientes con EB, particularmente durante las
exacerbaciones. La artritis afecta más comúnmente a las medianas y grandes
articulaciones, incluyendo la rodilla, tobillo, y muñeca. La inflamación se
hace evidente cuando se estudian muestras de líquido y tejido de biopsia
sinovial (255). En muchos pacientes, la artritis es intermitente, durando una a
tres semanas, aunque puede haber formas persistentes. Como ejemplo, en un
estudio de pacientes Griegos, la artritis fue pauciarticular en dos tercios de
los casos, monoarticular en un tercio, y poliarticular en menos del 5 por
ciento de los casos (216). Puede haber
sacroileitis, particularmente en pacientes con HLAB27, pero en algunos estudios
esta no es más probable que en pacientes sin EB (256). A pesar de su carácter
no erosivo, y no deformante un estudio usando un cuestionario (el multidimensional
Health Assessment Questionnaire), mostró que la artritis estuvo asociada con alteraciones funcionales similares a
pacientes con artritis reumatoidea (257).
Enfermedad renal.
La enfermedad renal en la EB es menos frecuente y a menudo menos
severa en otros tipos de vasculitis. Los pacientes con enfermedad renal pueden
tener proteinuria, hematuria o insuficiencia renal leve pero en algunos
pacientes puede evolucionar a fallo renal. El espectro de las enfermedades
renales fue ilustrado en un estudio de 159 pacientes con EB y compromiso renal.
La amiloidosis secundaria (AA) estuvo presente en 69, la glomerulonefritis en
51, la enfermedad vascular
(principalmente aneurismas de la arteria renal) en 35, y nefritis intersticial
en 4 (258). Entre los pacientes con glomerulonefritis hubo un espectro de
lesiones que iban desde nefropatía por IgA a glomerulonefritis con formación de
semilunas.
Los pacientes con amiloidosis (AA) se presentan típicamente con
síndrome nefrótico, o al menos con proteinuria significativa. En una serie de
14 casos el tiempo promedio entre el inicio de la EB hasta el inicio de la
nefropatía amiloide fue de ocho años (con un rango de 3 a 15 años) (259).
Enfermedad cardíaca.
La enfermedad cardíaca asintomática es infrecuente en EB. Las alteraciones
que pueden verse incluyen pericarditis, miocarditis, arteritis coronaria,
aneurismas septales auriculares, alteraciones del sistema de conducción,
arritmias ventriculares, endocarditis, fibrosis endomiocárdica, prolapso de
válvula mitral, e insuficiencia valvular (260-262). La aterosclerosis no parece
ocurrir de forma acelerada tal como dijimos antes, a diferencia de otras
enfermedades autoinmunes tales como lupus eritematoso sistémico (214).
Ulceraciones gastrointestinales.
Los pacientes pueden experimentar anorexia, náuseas, dolor abdominal,
o diarrea. Puede haber ulceraciones gastrointestinales en algunos pacientes con
EB, y puede verse aún perforaciones intestinales. Ulceraciones que son
generalmente discretas son vistas más a menudo en el ileon terminal, ciego, y
colon ascendente (Fig 21). Las úlceras orales ocurren frecuentemente en
pacientes con enfermedad inflamatoria intestinal (Crohn y colitis ulcerosa),
las cuales son indistinguibles de las de los pacientes con EB; por eso, la
enfermedad inflamatoria intestinal debe considerarse antes de realizarse el
diagnóstico de EB y viceversa.

Figura 21. Enfermedad de Behçet
comprometiendo el colon. Imagen magnificada de una radiografía de colon con doble
contraste (aire y bario), que muestra áreas segmentarias con úlceras (cabeza de
flecha), y pólipos inflamatorios (flecha)
Enfermedad pulmonar.
Además de las lesiones vasculares pulmonares ya discutidas en
enfermedad vascular, las alteraciones radiológicas incluyen pérdida de volumen
pulmonar, opacidades bien definidas, y sombras que pueden ser tanto nodulares
como reticulares. Aunque varios de estos hallazgos son infartos pulmonares, hemorragias, y
neumonía organizativas, no hay estudios que muestren correlación
radiológica/histopatológica (263). Otros hallazgos pulmonares misceláneos en EB
incluyen derrame pleural, arteritis pulmonar, o venulitis, estenosis bronquial,
abscesos, enfermedad obstructiva de la vía aérea, bronquitis crónica, y
fibrosis (247).
Embarazo.
La EB ha sido evaluada en ambarazo en muy pocos y pequeños estudios.
Un estudio caso control de 31 pacientes con 135 embarazos reportó más
remisiones que exacerbaciones durante el embarazo.
Las complicaciones fueron mayores en el grupo control que en el grupo
con EB, pero las complicaciones neonatales no fueron diferentes en los dos
grupos (264). Un análisis retrospectivo de la EB en 44 embarazos en 28
pacientes reportó remisión en 52,3 por ciento, exacerbación en 20,4 por ciento,
con tres abortos espontáneos (265).
Otros.
Los pacientes con EB pueden sufrir síndrome de repercusión del estado
general incluyendo fiebre y malestar general. Los problemas urinarios, y la
disfunción eréctil pueden ser consecuencia de enfermedad neural o vascular
(266). El compromiso del oído interno puede producir tinnitus o mareos (267). Puede
haber amiloidosis (259). Se ha descripto una posible asociación entre EB y
mielodisplasia (268,269). Un síndrome “pseudo-Behçet facticio” ha sido
descripto con compromiso predominante mucocutáneo.
La fibromialgia es frecuente en pacientes con EB. Un estudio de 70
pacientes con EB encontró que 37,1 por ciento de los pacientes reunían los
criterios del American College of
Rheumatology para fibromialgia. La fibromialgia se asoció a ansiedad y
depresión pero no con actividad de EB (270).
DIAGNÓSTICO.
No existe un test de laboratorio patognomónico que sea diagnóstico en
EB; como resultas de ello, el diagnóstico se hace en base a los hallazgos
clínicos. Los marcadores de inflamación tales como un nivel elevado de inmunocomplejos
circulantes, proteína C reactiva, y aumento de la velocidad de sedimentación
globular pueden estar elevados cuando la enfermedad está activa; sin embargo,
esos hallazgos son inespecíficos. Un número de alteraciones bioquímicas han
sido descriptas en EB y que van desde los hallazgos genéticos hasta un perfil y
nivel de citoquinas (ver patogenia de la EB). Seguramente en el futuro algunos
de ellos puedan ser útiles en el diagnóstico, pero las características de estos
hallazgos no son lo suficientemente definidos como para que sean utilizados hoy
con propósitos diagnósticos (71).
Varios criterios de clasificación han sido desarrollados para EB. Como
otros criterios de clasificación, esos criterios han sido desarrollados para
definir en forma operativa la enfermedad y facilitar ulteriores investigaciones
y educación médica, pero no han sido desarrollados para diagnóstico en casos
individuales (272-274). Tendrían estos criterios mayor precición en poblaciones
con baja prevalencia que en aquellas con alta prevalencia de EB (274).
Los antiguos criterios de Duffy, requerían aftas orales más al menos
dos de los siguientes: aftas genitales; sinovitis; uveítis posterior; patergia;
o meningoencefalitis, en ausencia de enfermedad inflamatoria intestinal o enfermedad
del colágeno (75).
Los nuevos criterios internacionales fueron publicados en 1990 (272).
Ellos requieren la presencia de aftas orales recurrentes (tres veces en un
años), más dos de los siguientes, en ausencia de otra enfermedad sistémica:
* Aftas genitales
recurrentes.
* Lesiones
oculares (incluyendo uveítis anterior o posterior, células en el humor
vítreo observadas con lámpara de hendidura, o vasculitis retiniana
observada por un oftalmólogo).
* Lesiones de
piel (incluyendo eritema nodoso, pseudovasculitis, lesiones
papulopustulares, o nódulos acneiformes compatibles con EB).
* Un test de
patergia positivo (una pápula de 2 mm o más que desarrolla en 24 a 48 hs después de la
inserción oblicua de una aguja calibre 20 a 25 en la piel,
generalmente en antebrazo).
Estos criterios parecen ser relativamente sencillos y específicos
(276,277).
La patergia es menos común en Europeos y Norteamericanos. Se ha
sugerido que la patergia podría ser sustituida por meningoencefalitis,
vasculitis cerebral, flebitis recurrente, arteritis, sinovitis, o ulceraciones
intestinales focales en estas poblaciones (276).
El retraso en el diagnóstico es común en áreas no endémicas, y el
diferimiento en el diagnóstico aumenta probablemente la morbi-mortalidad
(280,281).
DIAGNÓSTICO DIFERENCIAL.
El diagnóstico diferencial varía de acuerdo a cada hallazgo clínico
(282).
Las aftas orales están presentes en casi todos los pacientes con EB, y
el diagnóstico diferencial de úlceras orales recurrentes incluyen herpes
simplex, úlceras aftosas benignas, enfermedad inflamatoria intestinal, síndrome
de Stevens-Johnson, y otras enfermedades reumáticas sistémicas como lupus
eritematoso sistémico. El herpes puede ser descartado con una preparación de
Tzanck. Las prótesis dentarias y los productos para la higiene oral pueden
causar irritación o ulceración oral. Medicaciones como metotrexato pueden
causar úlceras orales. Otras causas de úlceras orales o estomatitis incluyen
penfigoide, pénfigo vulgar, penfigoide cicatrizal, liquen plano, y enfermedad
linear por IgA.
Otras causas de ulceraciones genitales como las infecciosas (HSV,
Treponema pallidum, Hemophilus ducreyi, Chlamydia trachomatis serovar L1-3
agente causal del linfogranuloma venéreo, y Klebsiella granulomatis, agente
causal del granuloma inguinal), y las causas no infecciosas como erupción fija
por drogas, neoplasias y trauma.
Muchas otras entidades pueden estar asociadas con vasculitis cutáneas
de pequeños vasos, enfermedad inflamatoria ocular, enfermedad neurológica,
enfermedad vascular, artritis, o enfermedad sistémica inexplicada. Ellas
incluyen: lupus eritematoso sistémico,
enfermedad inflamatoria intestinal, sarcoidosis, artritis reactivas, artritis
psoriásica, espondilitis anquilosante, artritis idiopática juvenil, fiebre
Mediterránea familiar y otros síndromes periódicos febriles, otras vasculitis,
esclerosis múltiple, infecciones sistémicas tales como tuberculosis, infección
por virus de la inmunodeficiencia humana, sífilis, y neoplasias.
PRONÓSTICO.
La EB tiene un curso remitente recidivante. La enfermedad parece ser
más severa en varones jóvenes (214,215). En una serie de 2200 pacientes con EB
en Corea sólo siete murieron de la enfermedad en un período de nueve años (283).
La mayor morbi/mortalidad se produce por causas neurológicas, oculares, y
enfermedad v vascular arterial o venosa (causando enfermedad pulmonar,
hemorragia digestiva, perforación gastrointestinal, y enfermedad
cerebrovascular).
La enfermedad mucocutánea, articular, y ocular son a menudo las
primeras manifestaciones durante los años iniciales de enfermedad, pero la
enfermedad de sistema nervioso central y la enfermedad de grandes vasos, si
desarrollan, típicamente lo hacen tardíamente en el curso de la enfermedad. Los
aneurismas de la arteria pulmonar y la hemoptisis asociada se asociaba a muy
mal pronóstico hasta hace unos años. Sin embargo, la sobrevida parece haber
mejorado, posiblemente como resultado del reconocimiento temprano y el
tratamiento usando glucocorticoides y otros inmunosupresores (284).
Las lesiones oculares y neurológicas pueden mejorar con
inmunosupresores pero a menudo no son totalmente reversibles. Sin terapia
agresiva, estas lesiones generalmente progresan. Una significativa proporción
de pacientes con enfermedad ocular, particularmente los descendientes de Turcos
o Japoneses, sufren pérdida progresiva de la visión.
TRATAMIENTO.
La literatura sobre el tratamiento de la EB consiste principalmente en
reporte de casos y pequeñas series de casos, con pocos estudios de seguimiento
que confirmen los reportes preliminares, y pocos ensayos randomizados.
En una revisión sistemática de la base de datos Cochrane de ensayos
conducidos hasta 1998, sólo 10 ensayos doble ciego reunían los criterios de
inclusión (284).
Los principales resultados del análisis de Cochrane arrojan alguna
duda de la eficacia de varios tratamientos clásicos de la EB incluyendo
colchicina, ciclofosfamida y glucocorticoides para la enfermedad ocular, y
colchicina para la artritis. Efectos
protectores del compromiso oftálmico fueron
confirmados tanto para ciclosporina como
Desde 1998 ha
habido sólo pequeños avances en la calidad de la literatura de la EB. Ellos
incluyenlos beneficios significativos en ensayos randomizados de colchicina en
enfermedad mucocutánea (285,286,287), y la introducción de la terapia con
anti-TNF-alfa.
Agentes Farmacológicos
Disponibles.
Los medicamentos empleados en la EB son enumerados más abajo.
Glucocorticoides.
Los glucocorticoides son la piedra angular del tratamiento de la EB
para pacientes con enfermedad moderada a severa. El compromiso severo de
algunos órganos puede requerir altas dosis; y dosis menores son utilizadas para
enfermedad menos severa (288,289). Algunos datos sugieren que la eficacia de
los glucocorticoides varía de acuerdo al régimen y la ruta de administración
(290). Las altas dosis en pulsos (metilprednisona 1 gramo administrado
intravenoso de uno a cinco días, más comúnmente tres días) se utiliza para
enfermedad severa con compromiso de órganos o compromiso de la vida.
Colchicina.
Esta droga ha sido usada desde hace mucho tiempo en EB,
particularmente para enfermedad mucocutánea. Los estudios clínicos con colchicina han mostrado resultados variables
(284,285,291,293). La terapia con
colchicina se asoció con reducción significativa en el número de articulaciones
con artritis en todos los pacientes y en úlceras genitales y eritema nodoso en
la mujer (285). Un estudios randomizado en 169 pacientes mostró mejoramiento
significativo en la actividad de la enfermedad, así como aftosis oral, aftosis
genital, psudofoliculitis, y eritema nodoso en pacientes tratados (294). Los
pacientes pueden desarrollar intolerancia gastrointestinal significativa a la
colchicina si la medicación es tomada en dosis mayores de 1,5 mg/día.
Azatioprina.
La azatioprina es una de las pocas medicaciones evaluadas en estudios
randomizados, doble ciego, controlado contra placebo en EB (295). En este
ensayo, 73 pacientes con EB fueron tratados con corticosteroides y azatioprina
(2,5 mg/día) o placebo. El seguimiento fue de dos años. Se demostró que el
grupo azatioprina tenía menos nuevo compromiso ocular, menos úlceras orales,
genitales y artritis.
Ciclofosfamida.
La ciclofosfamida
(1 a 2,5
mg/kg de peso/día, por vía oral; o 1
g , o 0,75
a 1 g/m2 por mes en infusión IV) es considerado un
agente efectivo para el compromiso neurológico de la EB.
Sin embargo
revisión de la base de datos Cochrane concluyó que había insuficiente evidencia
para el uso de ciclofosfamida en el tratamiento de la EB particularmente en sus
manifestaciones oculares (284).
Por otro lado, un
ensayo abierto comparó pulsos de ciclofosfamida contra pulsos de
metilprednisolona para uveítis, y encontró beneficios sólo con ciclofosfamida
(299), y algunos estudios observacionales han sugerido beneficios para la
enfermedad ocular y neurológica (300).
Ciclosporina.
La eficacia de la
ciclosporina en la EB ha sido evaluada inicialmente en la enfermedad ocular,
mucocutánea y articular. Alrededor dee la mitad de los pacientes con enfermedad
mucocutánea mejoraron (298,301,302).
La ciclosporina
generalmente se usa asociado a glucocorticoides (303) y a menudo en casos
severos o resistentes, con otros agentes inmunosupresores tales como
azatioprina (291). Muchos pacientes que respondieron a ciclosporina tienen
enfermedad recurrente después de discontinuar la terapia (301).
La ciclosporina
puede ser menos efectiva para el compromiso neurológico de la EB (287). Existen
guías (EULAR guidelines), que recomiendan precaución con el uso de ciclosporina
en pacientes con compromiso de sistema nervioso central (287).
Los efectos
adversos de la ciclosporina son comunes. Aproximadamente la mitad de los
pacientes tratados con esta droga presentan elevación de niveles de creatinina
que son reversibles cuando la droga es reducida (302). La hipertensión arterial
es común en el tratamiento con ciclosporina. La neurotoxicidad es vista
frecuentemente y puede ser difícil de diferenciar de la enfermedad neurológica
del la EB (306).
Tacrolimus, un
inhibidor de la calcineurina similar a ciclosporina tiene efectos similares
pero se tolera mejor (307).
Anti-TNF.
Los agentes que
inhiben las acciones del factor de necrosis tumoral (TNF)-alfa pueden mejorar
la actividad de la EB. Se han notado beneficios con infliximab y etanercept,
habiendo evidencias de que infliximab puede ser más efectivo (288,308).
Infliximab.
Un número de
estudios observacionales han sugerido la eficacia de infliximab para el
tratamiento de la enfermedad ocular, y de otros órganos en la EB (309,323,324).
Etanercept.
Etanercept puede
mejorar las manifestaciones mucosas y de piel de la EB (325).
Adalimumab.
Un número de
pequeños estudios han mostrado que el tratamiento con adalimumab puede ser
beneficioso (326,327,328).
Interferón-alfa.
Interferon
alfa-2ª e interferón alfa-2b (generalmente dados 3 a 19 millones de unidades
tres veces por semana) han mostrado beneficios en el tratamiento de la
enfermedad mucocutánea, ocular, articular, y neurológica en reporte de casos y
series de casos (329-339).
Micofenolato
mofetil.
Un ensayo con
micofenolato mofetil para enfermedad mucocutánea de EB fue interrumpido por
ausencia de eficacia en los primeros seis pacientes (346). Hay reporte de casos
que mostraron beneficio en algunos pacientes (347).
Talidomida.
Un ensayo clínico
ha sido llevado a cabo como monoterapia de EB (347) donde se demostró efecto
modesto a bueno aunque se han reportado efectos adversos como la aparición de
eritema nodoso y polineuropatía, por lo que su uso debe estar restringidos a
médicos con experiencia en su uso y al tratamiento de EB (348).
Rituximab.
En un ensayo randomizado
de 20 pacientes con vasculitis retiniana y edema resistente al tratamiento
citotóxico, el tratamiento con rituximab más metotrexato y prednisolona fue más
efectivo que el tratamiento con ciclofosfamida más azatioprina y prednisolona
(349).
Otros tratamientos.
Hay datos
limitados con el tratamiento con inmunoglobulinas intravenosas, plasmaféresis,
antibióticos, alemtuzumab, dapsona, antipalúdicos, transplante de stem cell
hemopoyética (autóloga o derivado de cordón umbilical) (331,350-362).
TRATAMIENTOS DE
ACUERDO A LOS SISTEMAS
ORGÁNICOS COMPROMETIDOS.
Artritis en EB.
La artritis no
deformante característica de la EB raramente es determinante del tipo de
terapia. Cuando se presentan síntomas articulares usualmente se presentan con
otras manifestaciones que son las que dictan la intensidad del tratamiento. Se
ha utilizado: colchicina 1 a
2 mg por día, AINES, glucocorticoides (prednisona 10 mg/día, azatioprina (295),
y metotrexato.
Manifestaciones
mucocutáneas de la EB.
Las
manifestaciones mucocutáneas de la EB incluyen aftas orales, úlceras genitales
(idénticas a las orales), y una variedad de otras lesiones de piel como
lesiones acneiformes, eritema nodoso, y pioderma gangrenoso.
Úlceras orales y
genitales.
Las úlceras
orales y genitales que son el sine qua non de la EB a menudo no responden a tratamientos tópicos y
requieren terapia sistémica (364). Los anestésicos tópicos son generalmente
inefectivos, y se sugiere para lesiones aisladas crema de acetonida de triamcinolona
aplicada tres a cuatro veces por día hasta que cese el dolor. El sucralfato
tópico 1g/5mL cuatro veces por día en forma de lavados orales, reduce el dolor
y acelera el tiempor de curación (360), lo mismo que el pimecrolimus (361).
Los corticoides
intralesionales de triamcinolona (5
a 10 mg/mL) son usados por dermatólogos.
La colchicina se
ha utilizado (285,292,293) pero puede producir intolerancia gastrointestinal
con dosis mayores de 1,5 mg/día.
Los corticoides
sistémicosse pueden usar en casos que no respondieron a tratamiento con corticoides
tópicos y colchicina, o cuando las lesiones son múltiples. En lesiones
mucocutáneas se comienza con 15 mg/día bajando hasta 10 mg/día después de una
semana. Cuando los requerimientos de corticoisteroides es de alta dosis o
períodos prolongados se puede usar azatioprina (295,296) 2,5 mg/kg/día, infliximab o etanercept,
ciclosporina (10 mg/kg/día) (365), e interferón-alfa (334). Talidomida
(347,366) se ha utilizado también.
Otras lesiones de
piel.
Con excepción de
eritema nodoso y pioderma gangrenoso, la mayoría de las manifestaciones
cutáneas de la EB (lesiones acneiformes,
y pápulopustulares, tromboflebitis superficiales, y púrpura palpable),
responden bien a medidas moderadas sobre todo colchicina (1 a 2 mg/día de colchicina) y
bajas a moderadas dosis de corticosteroides (hasta 40 mg de prednisona
inicialmente), para lesiones refractarias a colchicina. La dosis de
mantenimiento de 5 a
10 mg de prednisona usualmente es suficiente para controlar la mayoría de las
lesiones de piel de la EB.
El eritema nodoso
y el pioderma gangrenoso merecen una consideración especial.
El eritema
nodoso, a diferencia del asociado a sarcoidosis, enfemedad inflamatoria
intestinal y otras enfermedades inflamatorias, en la EB representa una
vasculitis de vasos de calibre mediano (367). Esta es la explicación de porqué
puede ulcerarse, y por eso hay que recurrir rápidamente a los corticosteroides
y otros inmunosupresores si la colchicina es inefectiva. La confirmación de la
presencia de vasculitis de vasos medianos en la biopsia, o la ulceración del
eritema nodoso es una indicación de glucocorticoides sistémicos combinados con
otro agente inmunosupresor. En este contexto se sugiere como terapia inicial
prednisona 40 a
60 mg/día y azatioprina 50 mg/día por vía oral. La dosis de prednisona se
mantiene por un mes y se comienza a descender en el término de tres a cuatro
meses. La azatioprinaAntes de iniciar azatioprina se aumenta mientras sea bien
tolerada en cuatro a seis semanas hasta una dosis de 2,5 mg/kg. Antes de iniciar
azatioprina se ha sugerido hacer un test genético para detectar la presencia de
metiltransferasa de tiopurina.
El pioderma
gangrenoso asociado a EB es a menudo complicado por el fenómeno de patergia (un
pinchazo con una aguja estéril puede evolucionar a una pústula cutánea en el
sitio del trauma, y el debridamiento de la lesión cutánea puede precipitar o
exacerbar el pioderma gangrenoso y producir expansión local. Por lo tanto el
debridamiento extenso del pioderma
gangrenoso es desaconsejado en EB.
Lesión ocular.
La lesión ocular
es tratada de acuerdo a su severidad (368). Las dos oculares manifestaciones más comunes, la uveítis
anterior y posterior tienen significativas diferencias terapéuticas. Sin
embargo, la uveítis anterior aislada es rara en EB. El escenario clínico más
común es la pan-uveítis.
Uveítis anterior.
La uveítis
anterior de la EB es tratada generalmente con corticosteroides tópicos y gotas
dilatadoras tales como escopolamina (0,25 por ciento) o ciclopentolato (1 por
ciento) para prevenir las sinequias y mantener la función pupilar. Si no se
usan midriáticos inicialmente se producen distorsiones pupilares causadas por
la formación de cicatrices o sinequias entre el iris y el cristalino.
Los pacientes
cuya uveítis mo es controlada con glucocorticoides tópics pueden requerir
tratamientos cortos con glucocorticoides sistémicos, por ejemplo prednisona 40
mg por día vía oral.
Uveítis posterior
y otras manifestciones mayores de enfermedad.
La uveítis
posterior (inflamación en el tracto uveal posterior al cristalino) en la EB
constituye un serio problema en términos de visión y requiere inmunosupresión
intensiva. La combinación de altas dosis de corticosteroides y otros agentes
inmunosupresores (usualmente azatioprina) son generalmente requeridos.
El uso de
corticosteroides en la uveítis posterior ayuda a reducir la inflamación aguda,
pero si no se usan combinados a otros inmunosupresores la actividad de la
enfermedad tiende a recurrir. Generalmente se comienza con prednisona 1 mg/kg
de peso por día durante un mes y se comienza a disminuir la dosis. A veces se
usa inicialmente metilprednisona en pulsos de 1 g/tres días (288,289,369). Se
puede inyectar intraocularmente triamcinolona que es útil en la panuveítis
durante dos a seis meses(367). Los glucocorticoides se usan asociados
usualmente a azatioprina.
Los agentes
inmunosupresores utilizados como agentes asociados a corticosteroides son
azatioprina, infliximab y ciclosporina en la uveítis posterior. No hay estudios
que demuestren superioridad de una droga con respecto a otra.
En casos
refractarios se ha utilizado interferon alfa-2a (343,344). Pacientes con
vasculitis retiniana o enfermedad ocular refractaria a la combinación de
corticosteroides, ciclosporina y azatioprina pueden responder con interferón
alfa-2a sólo (336,340,371,372).
Rituximab ha sido
beneficioso en pequeños ensayos con severas manifestaciones oculares
resistentes a los citotóxicos (349).
Ciclofosfamida puede
ser empleada en casos refractarios en los que la actividad de la enfermedad
amerita y justifica los riesgos de su uso.
Finalmente
metotrexato, micofenolato mofetil y otros agentes anti-TNF pueden ser
considerados a pesar de la pequeña cantidad de ensayos publicados.
Enfermedad
neurológica.
Como con la
enfermedad ocular hay un rango de severidad de enfermedad entre las lesiones
neurológicas. Las lesiones focales parenquimatosas, las encefalitis, y las
vasculitis de vasos de mediano calibre son todas enfermedades que ponen en
riesgo potencialmente la vida, que deben ser tratados de la misma manera que la
uveítis posterior.
Las meningitis
asépticas pueden ocurrir intermitentemente en pacientes con EB. En ausencia de
otras manifestaciones de enfermedad que requieran terapia más intensiva, las
mayorías de las meningitis asépticas responden a altas dosis de
glucocorticoides (1 mg/kg/día hasta un máximo de 80 mg/día). Las recuerrencias
se tratan asociando otros inmunosupresores como azatioprina.
El tratamiento de
la trombosis de los senos cerebrales es tratado más abajo.
Enfermedad
gastrointestinal.
Las pequeñas
ulceraciones observadas en el ileon, en el ciego, y colon ascendente son
comunes. La presencia de compromiso significativo gastrointestinal señala la
necesidad de terapia con glucocorticoides más un agente adicional para evitar
uso a largo plazo de prednisona.
Para las úlceras
gastrointestinales en EB se sugiere glucocorticoides (o,5 a 1 mg por kg/día
(373) más azatioprina 2,5 mg/kg/día.
Hay reportes que
hablan de infliximab en forma similar a la utilizada en enfermedad inflamatoria
intestinal (374,375), y la sulfasalazina pueden utilizarse con cierto éxito.
Enfermedad renal.
Aunque las
alteraciones urinarias (proteinuria y/o hematuria) ocurren en aproximadamente
10 por ciento de los pacientes, las lesions renales son raras (376). El
espectro de las enfermedades renales asociadas
en una revisión de 159 pacientes con EB se vio amiloidosis secundaria
(AA) en 69, glomerulonefritis en 51, enfermedad vascular (principalmente aneurismas
de la artria renal en 35, y nefritis intersticial en 4 (377). Entre los
pacientes con glomerulonefritis hubo un espectro de lesiones que iban desde la
nefropatía por IgA, hasta glomerulonefritis con formación de semilunas.
El tratamiento de
la enfermedad de Behçet renal varía con la lesión de base. Para la nefritis
mínima o leve (hematuria, excreción de menos de 500 mg a 1000 mg de proteínas
por día y creatinina normal) pueden no requerir terapia específica. Los
pacienets con amiloidosis que resulta de la inflamación crónica, requieren tratamiento
de la EB. La terapia preferida es colchicina. El tratamiento de los aneurismas
de la arteria renal se discuten en el próximo apartado.
Respecto a las
glomerulonefritis clínicamente importantes hay pocas series de casos como para
guiar la terapia en pacientes con EB. Dado la naturaleza vasculítica de la EB
es razonable extrapolar los mediacamentos usados en enfermedad de Wegener,
particularmente en pacientes con lesiones necrotizantes focales o
glomerulonefritis rápidamente evolutivas con formación de semilunas.
Enfermedad de
arterias grandes.
El compromiso
arterial en la EB es infrecuente pero cuando se produce puede conducir a
formación de aneurismas en arterias de mediano o gran tamaño (377,378). En una
revisión de 159 pacientes citados más arriba con compromiso renal, 35 tuvieron
enfermedad vascular, la mayoría de ellos aneurismas de la arteria renal (377).
Esta complicación puede requerir tratamientos combinados médicos y quirúrgicos
o tratamientos por radiología intervencionista.
El approach
médico incluye altas dosis de corticosteroides y otros agentes
inmunosupresores, típicamente ciclofosfamida (206).
Como consecuencia
del tratamiento de estas complicaciones vasculares se ha observado que altas
dosis de corticosteroides no mejoran la enfermedad oclusiva y pueden contribuir
a infecciones fatales. Los aneurismas tienen peor pronóstico que la enfermedad
oclusiva. La reparación quirúrgica es necesaria para prevenir la ruptura de los
aneurismas, y es necesaria la terapia inmunosupresora en el posoperatorio. Los
glucocorticoides más otros inmunosupresores parecen ser más efetcivos que los
corticoides solos. La anticoagulación se empló exitosamente en varios casos
empíricamente para prevenir trombosis del injerto en el posoperatorio.
Las complicaciones
vasculares siguiendo la cirugía de bypass es común en pacientes con EB. Los más
frecuente es la oclusión del injerto con formación de aneurismas en el sitio de
anastomosis (382).
La radiología
intervencionista es una alternativa para pacientes con dilatación aneurismática
de la aorta de y de las grandes arterias (383,384) La colocación percutánea de
stents fue exitosa en seis de siete pacientes con aneurismas arteriales
8aóticos, subclavios, carotídeos, braquiocefálicos e ilíacos) (383).
Trombosis venosas.
La enfermedad
venosa en la EB es considerada el resultado de la inflamción endotelial que
conduce a trombosis (206, 385). El approach para prevenir eventos trombóticos
en EB es el control de la inflamación, más que la institución de una terapia anticoagulante.
Sin embargo, si ocurre trombosis venosa hay que anticoagular a dosis standard.
Las trombosis
venosas complicadas con eventos tales como trombosis de los senos venosos
cerebrales, trombosis venosas cerebrales, y trombosis en la vena pulmonar pueden
ocurrir en EB.
El tratamiento
consiste en controlar la inflamación sistémica asociada a la EB,
anticoagulación (generlamente con warfarina), y en algunos casos trombolisis
local. Las convulsiones y la hipertensión intracraneana pueden ser consecuencia
de trombosis venosa cerebral requiriendo otras intervenciones (386).
Referencias
Bibliográficas.
18.
Sakane T, Takeno M. Novel approaches to Behçet's disease. Expert Opin Investig Drugs
2000; 9:1993.
101.
Yato, H, Matsumoto, Y.
CD56+ T cells in peripheral blood of uveitis patients. Br J Ophthalmol 199; 83:1386.
211) O'Duffy JD. Behcet's syndrome. In: Primer on the
Rheumatic Diseases, 10th, Arthritis Foundation, Atlanta
240) Lee SH, Yoon PH, Park SJ, Kim DI. MRI findings in neuro-behçet's disease. Clin Radiol
2001; 56:485.
245) Koç Y, Güllü I, Akpek G, et al. Vascular involvement in Behçet's disease.
J Rheumatol 1992;
19:402.
280) Alpsoy E. Behçet's disease: treatment of mucocutaneous lesions. Clin Exp Rheumatol 2005;
23:532.
306)Sakane T, Takeno M. Novel approaches to Behçet's disease. Expert Opin Investig Drugs
2000; 9:1993.
361)Tellier Z. Intravenous immunoglobulin in eye involvement. Clin Rev Allergy Immunol 2005;
29:295.
363)Alpsoy E. Behçet's disease: treatment of mucocutaneous lesions. Clin Exp Rheumatol 2005;
23:532.




