
Paciente femenina de 47 años
Motivo de consulta:
Dolores generalizados, pérdida de fuerzas, rash cutáneo.
Enfermedad actual:
Dice que hace 45 días le diagnosticaron síndrome gripal, a raiz de una consulta telefónica hecha por mialgias y rash cutáneo en plena epidemia de gripe A H1N1. Simultaneamente, su hijo cursó un episodio de gripe por lo que se le aconsejó a ella, reposo y analgésicos.
Desde entonces refiere pérdida de fuerzas generalizadas que le impiden su trabajo habitual de maestra rural, las que continúa realizando a expensas de un gran esfuerzo. En los últimos 30 días comenzó a notar dificultad en la marcha por debilidad y dolor en miembros inferiores, sobre todo a nivel d e muslo. También refiere dolor en cintura escapular e imposibilidad de llevar a cabo tareas que demanden esfuerzo con los brazos y más recientemente, notó dificultad para elevar sus brazos, lo que dificulta maniobras como por ejemplo peinarse.
e muslo. También refiere dolor en cintura escapular e imposibilidad de llevar a cabo tareas que demanden esfuerzo con los brazos y más recientemente, notó dificultad para elevar sus brazos, lo que dificulta maniobras como por ejemplo peinarse.
El rash cutáneo afecta la cara, donde tiene compromiso malar bilateral (eritema en vespertilio o “butterfly rash”) y compromiso en frente. Refiere fotosensibilidad. También refiere rash intensamente pruriginoso en cuero cabelludo, cuello, en raiz de muslos cara lateral externa, y región lumbar baja.
Durante este tiempo no refiere haber presentado fiebre, escalofrios, sudoración nocturna ni otros síntomas que orienten hacia determinado sistema. No refiere síndrome de Raynaud. No adelgazó. No hay un predominio horario para su sintomatología.
En los últimos 20 días nota disnea significativa de esfuerzo clase funcional 2/3, de la que se repone al cesar su esfuerzo, y últimamente dificultad para tragar, presentando durante la deglución, crisis de tos y disnea, por lo que decide consultar.
Antecedentes de enfermedad actual:
Motivo de consulta:
Dolores generalizados, pérdida de fuerzas, rash cutáneo.
Enfermedad actual:
Dice que hace 45 días le diagnosticaron síndrome gripal, a raiz de una consulta telefónica hecha por mialgias y rash cutáneo en plena epidemia de gripe A H1N1. Simultaneamente, su hijo cursó un episodio de gripe por lo que se le aconsejó a ella, reposo y analgésicos.
Desde entonces refiere pérdida de fuerzas generalizadas que le impiden su trabajo habitual de maestra rural, las que continúa realizando a expensas de un gran esfuerzo. En los últimos 30 días comenzó a notar dificultad en la marcha por debilidad y dolor en miembros inferiores, sobre todo a nivel d
 e muslo. También refiere dolor en cintura escapular e imposibilidad de llevar a cabo tareas que demanden esfuerzo con los brazos y más recientemente, notó dificultad para elevar sus brazos, lo que dificulta maniobras como por ejemplo peinarse.
e muslo. También refiere dolor en cintura escapular e imposibilidad de llevar a cabo tareas que demanden esfuerzo con los brazos y más recientemente, notó dificultad para elevar sus brazos, lo que dificulta maniobras como por ejemplo peinarse.El rash cutáneo afecta la cara, donde tiene compromiso malar bilateral (eritema en vespertilio o “butterfly rash”) y compromiso en frente. Refiere fotosensibilidad. También refiere rash intensamente pruriginoso en cuero cabelludo, cuello, en raiz de muslos cara lateral externa, y región lumbar baja.
Durante este tiempo no refiere haber presentado fiebre, escalofrios, sudoración nocturna ni otros síntomas que orienten hacia determinado sistema. No refiere síndrome de Raynaud. No adelgazó. No hay un predominio horario para su sintomatología.
En los últimos 20 días nota disnea significativa de esfuerzo clase funcional 2/3, de la que se repone al cesar su esfuerzo, y últimamente dificultad para tragar, presentando durante la deglución, crisis de tos y disnea, por lo que decide consultar.
Antecedentes de enfermedad actual:

Según la paciente hace más de 1 año que nota pérdida de fuerzas para realizar esfuerzos moderados como llevar un peso de 2 o 3 kg y caminar con él durante 100 metros, habiendo llegado “agotada” en varias oportunidades a su casa después de realizar las compras de rutina. De esta sintomatología se recuperaba después de descansar unos minutos.
Antecedentes personales:
Rinitis estacional, sinusitis alérgica. Dos cesáreas. Dos hijos
 vivos y sanos.
vivos y sanos.No fuma, no ingiere bebidas alcohólicas, ni ha usado drogas.
Antecedentes familiares:
Padre asmático. Madre con malformación cardíaca congénita. HTA.
Hermanos 2. Una hermana con psoriasis, y un hermano con parálisis facial periférica.
Examen físico:
Hermanos 2. Una hermana con psoriasis, y un hermano con parálisis facial periférica.
Examen físico:
Paciente en mal estado general, con severa dificultad para movilizarse, le cuesta levantarse de la silla y tiene una marcha lenta y dificultosa con ayuda de otra persona.
Afebril, TA 110/80, fcia cardíaca 100 por minuto.
Afebril, TA 110/80, fcia cardíaca 100 por minuto.
Piel:
Se observa un tenue rash cutáneo en frente y en ambas regiones malares que desaparecen a la vitropresión. Rash eritematoso en el cuello, en ambas regiones laterales externas de muslo a nivel de la raiz del miembro inferior, y en región lumbar baja, todos ellos con lesiones de rascado, por intenso pr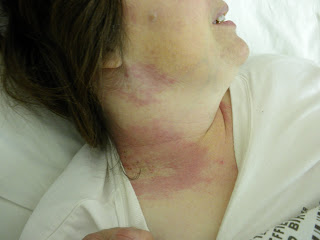 urito. En cuero cabelludo, donde refiere prurito, se palpan lesiones ligeramente elevadas con trauma por rascado e impetiginización sobreagregada. No hay alopecía.
urito. En cuero cabelludo, donde refiere prurito, se palpan lesiones ligeramente elevadas con trauma por rascado e impetiginización sobreagregada. No hay alopecía.
 urito. En cuero cabelludo, donde refiere prurito, se palpan lesiones ligeramente elevadas con trauma por rascado e impetiginización sobreagregada. No hay alopecía.
urito. En cuero cabelludo, donde refiere prurito, se palpan lesiones ligeramente elevadas con trauma por rascado e impetiginización sobreagregada. No hay alopecía. Tejido celular subcutáneo:
Llama la atención un importante edema duro, generalizado que no deja godet franco, y que predomina en brazos, antebrazos, y región lateral del cuello, y que recuerda al del hipotiroidismo (mixedema, mucinosis cutánea).
Mucosas:
Se observan en región gingival inferior y piso de la boca lesiones ulcerosas en número de dos de 5 mm intensamente dolorosas.
El resto del examen de la boca es normal, con buena movilidad del paladar blando, pilares amigdalinos, y lengua. Tiene dificultad para abrir completamente la boca, no sabe si es por dolor.
La paciente refiere odinofagia, y su voz tiene un tono nasal. Sialorrea.
El resto del examen de la boca es normal, con buena movilidad del paladar blando, pilares amigdalinos, y lengua. Tiene dificultad para abrir completamente la boca, no sabe si es por dolor.
La paciente refiere odinofagia, y su voz tiene un tono nasal. Sialorrea.
Aparato locomotor:
Se observa una pérdida de fuerzas generalizadas, a predominio de miembros inferiores y superiores proximal. El compromiso es más evidente en cuádriceps y deltoides, con casi imposibilidad de levantarse de la silla sin ayuda de los brazos. Imposibilidad de la abeducción de miembros superiores.
La palpación de los músculos del cuello y brazos es dolorosa, así como a nivel de muslos. Pérdida de fuerzas en la flexión anterior, posterior, y lateralización de la cabeza, notando que no puede mantener la cabeza erguida por mucho tiempo. Cuando permanece sentada, la cabeza tiene tendencia a caer hacia atrás o hacia adelante con intenso dolor, que hace que rehuse estar en dicha posición por mucho tiempo. Solicita que se le sostenga la cabeza en los cambios de postura de acostada a sentada por la anormal movilidad cefálica, y el dolor que esto provoca en el intento de hacerlo.
La palpación de los músculos del cuello y brazos es dolorosa, así como a nivel de muslos. Pérdida de fuerzas en la flexión anterior, posterior, y lateralización de la cabeza, notando que no puede mantener la cabeza erguida por mucho tiempo. Cuando permanece sentada, la cabeza tiene tendencia a caer hacia atrás o hacia adelante con intenso dolor, que hace que rehuse estar en dicha posición por mucho tiempo. Solicita que se le sostenga la cabeza en los cambios de postura de acostada a sentada por la anormal movilidad cefálica, y el dolor que esto provoca en el intento de hacerlo.
Aparato respiratorio:
Buena entrada de aire bilateral. Se auscultan algunos roncus y secreciones gruesas, a predominio de base pulmonar izquierda, que son aclaradas con la tos.
Aparato cardiovascular:
Taquicardia sinusal. Normotensa.
`
Abdomen: sp
Sistema nervioso:
Sensibilidad conservada, fuerza muscular descripta arriba francamente disminuída. Reflejos osteotendinosos disminuidos por probable compromiso miopático
No hay fasciculaciones.
Laboratorio:
No hay fasciculaciones.
Laboratorio:
GR 3.770.000 Hto 34%, Hb 11,4 g/dl, GB 11300 fórmula conservada, Plaquetas 189.000. Eritrosedimentación 32 mm/hora. Urea 36 mg/dl, creatinina 0,60 mg/dl. TGP 120 U/L (Normal hasta 48) TGO 182 U/L (Normal hasta 48), resto de hepatograma normal. Aldolasa 24,0 U/L (Normal hasta 7,6) Colesterol 320 mg/dl. Ionograma normal.
TSH 2,94.
FAN (+) 1/160.
FAN (+) 1/160.
Sedimento urinario: normal.
CPK 3712 U/L (Valor normal hasta 80)
Tele Rx de tórax: normal.
Rx de partes blandas: no se observa calcinosis.
Examen ginecológico, PAP, y mamografía: normales.
Espirometría:
CVF 2,71 (93%) 
VEF1 seg 2,18 (89%)
Pico espiratorio flujo 2,8 (50%)

VEF1 seg 2,18 (89%)
Pico espiratorio flujo 2,8 (50%)
Resonancia magnética de cintura escapular:
Se observa líquido laminar subdérmico rodeando ambos músculos deltoides a predominio izquierdo. Existe también líquido entre el plano muscular supraespinoso y deltoides de ambos hombros. Se aprecian cambios internos de señal de fibras de la porción lateral y central de la masa muscular del deltoides derecho (miolisis). La región subescapular bilateral también demuestra líquido laminar en distribución subaponeurótica.
cambios internos de señal de fibras de la porción lateral y central de la masa muscular del deltoides derecho (miolisis). La región subescapular bilateral también demuestra líquido laminar en distribución subaponeurótica.
Se observa líquido laminar subdérmico rodeando ambos músculos deltoides a predominio izquierdo. Existe también líquido entre el plano muscular supraespinoso y deltoides de ambos hombros. Se aprecian
 cambios internos de señal de fibras de la porción lateral y central de la masa muscular del deltoides derecho (miolisis). La región subescapular bilateral también demuestra líquido laminar en distribución subaponeurótica.
cambios internos de señal de fibras de la porción lateral y central de la masa muscular del deltoides derecho (miolisis). La región subescapular bilateral también demuestra líquido laminar en distribución subaponeurótica. Biopsia de deltoides izquierdo:
Estructura muscular estriada con infiltrados linfocitarios leves de endomisio y perimisio.
Estructura muscular estriada con infiltrados linfocitarios leves de endomisio y perimisio.
Biopsia de piel:
Estructura epidérmica conservada con áreas focales y paraqueratósicas de infiltrados linfocíticos de la dermis papilar perivascular y perianexial.
Ambos hallazgos configuran un cuadro de dermatomiositis.
Video deglución:
Mediante ingesta de semisólidos con el agregado de sustancia de contraste (vista lateral), se observa dificultad en primer tiempo deglutorio. Disfunción lingual, caída precoz del alimento. Retención en el esfínter esofágico superior. Con líquido se observa correcto desplazamiento del contenido.
La paciente requiere ejercitar la maniobra de chin-down para compensar deglución. Se sugiere realizar tratamiento fonoaudiológico para desarrollo de praxias orofaciales y pautas de alimentación cuando su estado lo permita.
No se pudo realizar estudio electromiográfico por carecer en nuestro hospital de equipamiento para dicho test, y no estar la paciente en condiciones de ser trasladada para realizarlo.
Estructura epidérmica conservada con áreas focales y paraqueratósicas de infiltrados linfocíticos de la dermis papilar perivascular y perianexial.
Ambos hallazgos configuran un cuadro de dermatomiositis.
Video deglución:
Mediante ingesta de semisólidos con el agregado de sustancia de contraste (vista lateral), se observa dificultad en primer tiempo deglutorio. Disfunción lingual, caída precoz del alimento. Retención en el esfínter esofágico superior. Con líquido se observa correcto desplazamiento del contenido.
La paciente requiere ejercitar la maniobra de chin-down para compensar deglución. Se sugiere realizar tratamiento fonoaudiológico para desarrollo de praxias orofaciales y pautas de alimentación cuando su estado lo permita.
No se pudo realizar estudio electromiográfico por carecer en nuestro hospital de equipamiento para dicho test, y no estar la paciente en condiciones de ser trasladada para realizarlo.
TAC abdomino-pelviana:
Normal.
Se estableció diagnóstico de dermatomiositis.
Normal.
Se estableció diagnóstico de dermatomiositis.
Tratamiento:
Se colocó sonda nasogástrica para alimentación.
Se hicieron tres días de pulsos con 40 mg de dexametasona, y al cuarto día comenzó con:
Metilprednisona 60 mg/día.
Metotrexato 15 mg/semana.
Bactrim fuerte 1 comprimido por día.
Clonazepan 0,5 mg 2 veces por día.
Tuvo buena tolerancia al tratamiento y buena respuesta clínica al cabo de 15 días, objetivándose mejoría en la motilidad y la fuerza muscular en cintura escapular y pelviana, así como en los grupos musculares del cuello con recuperación de la capacidad de mantener la cabeza erguida, como también de elevar los brazos para realizar movimientos como peinarse. Recuperó la capacidad de rotar su cuerpo en la cama así como mantenerse sentada sin ayuda por varias horas. Se ha objetivado capacidad de caminar con menor dificultad, con ayuda, y de movilizarse hasta el baño.
Hubo una notable mejoría en las lesiones dermatológicas sin necesidad de recurrir a tratamientos más específicos (antipalúdicos). Desapareció el prurito y las lesiones han disminuido la intensidad del eritema, persistiendo una pálida erupción macular en región lumbar y región lateral de cuello. Desaparecieron las lesiones cutáneas faciales.
También se observó una notable mejoría del edema duro (de aspecto mixedematoso) que presentaba en ambos brazos, en cuello y muslos.
La desaparición de este edema infiltrativo, puso en evidencia la intensa atrofia muscular generalizada que presenta actualmente la paciente, a pesar de la mejoría de su fuerza. El tratamiento de rehabilitación con ejercicios activos de fortalecimiento muscular, fue diferido por indicación reumatológica, hasta tener más controlado el proceso inflamatorio.
A pesar de la mejoría general de la paciente persiste la dificultad deglutoria, con mal manejo de secreciones, sialorrea y odinofagia por lo que se solicita estudio endoscópico de vía aérea superior y cavum, por la posibilidad de lesión orgánica a ese nivel, no visualizado en el estudio de video-deglución.
Al cabo de 20 días de tratamiento se realizó dosaje de CPK de control obteniéndose un resultado de 237 U/L (anterior 3712 U/L).
Se hicieron tres días de pulsos con 40 mg de dexametasona, y al cuarto día comenzó con:
Metilprednisona 60 mg/día.
Metotrexato 15 mg/semana.
Bactrim fuerte 1 comprimido por día.
Clonazepan 0,5 mg 2 veces por día.
Tuvo buena tolerancia al tratamiento y buena respuesta clínica al cabo de 15 días, objetivándose mejoría en la motilidad y la fuerza muscular en cintura escapular y pelviana, así como en los grupos musculares del cuello con recuperación de la capacidad de mantener la cabeza erguida, como también de elevar los brazos para realizar movimientos como peinarse. Recuperó la capacidad de rotar su cuerpo en la cama así como mantenerse sentada sin ayuda por varias horas. Se ha objetivado capacidad de caminar con menor dificultad, con ayuda, y de movilizarse hasta el baño.
Hubo una notable mejoría en las lesiones dermatológicas sin necesidad de recurrir a tratamientos más específicos (antipalúdicos). Desapareció el prurito y las lesiones han disminuido la intensidad del eritema, persistiendo una pálida erupción macular en región lumbar y región lateral de cuello. Desaparecieron las lesiones cutáneas faciales.
También se observó una notable mejoría del edema duro (de aspecto mixedematoso) que presentaba en ambos brazos, en cuello y muslos.
La desaparición de este edema infiltrativo, puso en evidencia la intensa atrofia muscular generalizada que presenta actualmente la paciente, a pesar de la mejoría de su fuerza. El tratamiento de rehabilitación con ejercicios activos de fortalecimiento muscular, fue diferido por indicación reumatológica, hasta tener más controlado el proceso inflamatorio.
A pesar de la mejoría general de la paciente persiste la dificultad deglutoria, con mal manejo de secreciones, sialorrea y odinofagia por lo que se solicita estudio endoscópico de vía aérea superior y cavum, por la posibilidad de lesión orgánica a ese nivel, no visualizado en el estudio de video-deglución.
Al cabo de 20 días de tratamiento se realizó dosaje de CPK de control obteniéndose un resultado de 237 U/L (anterior 3712 U/L).
Estudios pendientes:
Anti-Jo1. Anti SRP. Anti-Mi-2.
Fibroendoscopía de cavum y vías aéreas superiores
Manifestaciones clínicas y diagnóstico de la dermatomiositis (DM) y la polimiositis (PM) en adultos.
Manifestaciones clínicas y diagnóstico de la dermatomiositis (DM) y la polimiositis (PM) en adultos.
Introducción:
La dermatomiositis (DM) y la polimiositis (PM) son clasificadas como miopatías inflamatorias (1-3) La prevalencia de ambos trastornos
 es de 1/100.000 en la población general. Hay una predilección por el sexo femenino en una relación de 2/1. El pico de incidencia en adultos ocurre entre los 40 y los 50 años, pero puede comenzar a cualquier edad. (4-5)>
es de 1/100.000 en la población general. Hay una predilección por el sexo femenino en una relación de 2/1. El pico de incidencia en adultos ocurre entre los 40 y los 50 años, pero puede comenzar a cualquier edad. (4-5)>Diferencias entre la DM y la PM
Aunque la DM y la PM comparten la debilidad muscular, hay diferencias tanto clínicas como patogénicas entre ambos trastornos:
La DM se asocia a una variedad de manifestaciones en piel, incluyendo el signo de Gottron (piel eritematosa, engrosada y descamativa en los nudillos), el signo del manto o del chal (lesiones eritematosas en la “V” del cuello y en la parte anterior y posterior del tórax así como los hombros, justamente en la zonas
que cubre un chal), el exantema en heliotropo (coloración lila o violácea en los párpados), y la eritrodermia generalizada.
La DM se asocia más probablemente a cáncer que la PM.
La DM se caracteriza por deposición de inmunocomplejos en los vasos y es considerada en parte, como una vasculopatía mediada por complemento. En contraste, la PM parece reflejar la injuria muscular directa, mediadas por células-T.
Criterios de clasificación:
>Varios sets de clasificación han sido desarrollados para clasificar la DM y la PM.
Criterios de Bohan y Peter
Los criterios originales de Bohan y Peter formulados en 1975 incluyen los siguientes elementos:
1) Debilidad muscular proximal simétrica.
2) Rash típico de DM, que es el único hallazgo distintivo entre la DM y la PM.
3) Enzimas musculares elevadas.
4) Cambios miopáticos en la electromiografía.
5) Hallazgos característicos en la biopsia muscular, y ausencia de signos histopatológicos de otras miopatías.
Estos criterios fueron formulados antes de existir los autoanticuerpos específicos de miositis (ver más abajo). Además, la miositis por cuerpos de inclusión, que comparte hallazgos clínicos con DM y PM, no se conoció hasta 1980.
Por lo tanto, pacientes diagnosticados de DM y PM con estos criterios, pueden tener otros trastornos. Muchas enfermedades clasificadas como PM con los criterios actuales se clasifican como miositis de superposición (“overlap” myositis) (9)
Esquemas alternativos de clasificación
Dos nuevos esquemas de clasificación alternativos han aparecido desde 2004 (9,10). Los nuevos dos sistemas utilizan poblaciones significativamente diferentes. Además una clasificación se basa principalmente en criterios histológicos mientras que la otra se basa principalmente en elementos clínicos.
Un approach, la clasifica de acuerdo a elementos “clínico-serológicos” específicamente el testeo de autoanticuerpos. Excluyendo la miositis por cuerpos de inclusión, 4 categorías de miopatías inflamatorias fueron reconocidas:
1) PM pura.
2) DM pura.
3) Miositis de superposición, que fue definida como miositis más cualquier hallazgo clínico de superposición aparte del rash, y/cualquier autoanticuerpo de superposición. (ver más abajo).
4) Miositis asociada a cáncer, que fue definida como una miositis con elementos clínicos paraneoplásicos, y sin autoanticuerpos de superposición o anticuerpos anti-Mi-2.
El segundo esquema propuesto por un workshop de expertos, excluye las miositis asociadas a otras enfermedades del tejido conectivo o enfermedades malignas, pero incluye las siguientes categorías (10):
>1) Miositis por cuerpos de inclusión.
2) PM definitiva.
3) PM probable.
4) DM definitiva.
5) DM probable.
6) DM amiopática, también llamada dermatomiositis sin miositis.
7) Posible DM sin dermatitis.
8) Miositis inespecífica.
9) Miopatía necrotizante inmuno-mediada.
Estas dos categorías de clasificación están basadas en criterios clínicos, histopatológicos, y hallazgos de laboratorio, incluyendo determinación de autoanticuerpos.
Estas dos nuevas clasificaciones permiten mayor precisión diagnóstica, y predecir el curso de la enfermedad y la respuesta al tratamiento que la antigua clasificación de Bohan y Peter.>
Inmunopatogénesis:
Aunque los hallazgos histológicos tanto de DM como de PM incluyen: necrosis de fibras musculares, degeneración y regeneración, y un infiltrado celular inflamatorio, ciertos hallazgos característicos en esas dos diferentes enfermedades, reflejan diferentes vías patofisiológicas:
· La DM es considerada un trastorno mediado-humoralmente, en el que el infiltrado celular, localizado principalmente en la región perifascicular, está focalizado alrededor de los vasos (Figura 1)(12-14).

Figura 1. Tinción con hematoxilina-eosina (x20) de una biopsia muscular de un paciente con DM, mostrando inflamación perivascular y perimisial así como necrosis perifascicular.
(Fuente: UpToDate)
El complejo de ataque de la membrana del complemento (MAC) (C5b-C9) es detectable en la pared de los vasos (Figura 2) antes de la aparición del infiltrado inflamatorio en la DM pero no en la PM. (12)

Figura 2: Biopsia muscular en un paciente con DM mostrando deposición de C5b-9 en los vasos del perimisio.
(Fuente UpToDate)

Figura 2: Biopsia muscular en un paciente con DM mostrando deposición de C5b-9 en los vasos del perimisio.
(Fuente UpToDate)
Aunque la DM está claramente asociada a una vasculopatía en que participa el complemento, no se sabe aún si esta vasculopatía es mediada únicamente por complemento, o si la deposición de proteínas del complemento y otros complejos inmunes, es secundaria a otros eventos patofisiológicos. (15,16)
El infiltrado inflamatorio está compuesto por células B y células dendrítico-plasmocitoides CD4+. Otros hallazgos típicos incluyen atrofia perifascicular y fibrosis. Las fibras musculares anormales están usualmente agrupadas en una porción del fascículo, sugestivas de microinfartos mediados por alteración vascular (12).
· En la PM, el infiltrado celular está predominatemente dentro del fascículo con células inflamatorias que invaden las fibras musculares (Figura 3) (14).
 Figura 3: En el panel de la izquierda y en derecha se ven biopsias en PM. Hay un intenso infiltrado mononuclear intersticial con alguna degeneración de miocitos.
Figura 3: En el panel de la izquierda y en derecha se ven biopsias en PM. Hay un intenso infiltrado mononuclear intersticial con alguna degeneración de miocitos.
(Fuente UpToDate)
El infiltrado inflamatorio está compuesto por células B y células dendrítico-plasmocitoides CD4+. Otros hallazgos típicos incluyen atrofia perifascicular y fibrosis. Las fibras musculares anormales están usualmente agrupadas en una porción del fascículo, sugestivas de microinfartos mediados por alteración vascular (12).
· En la PM, el infiltrado celular está predominatemente dentro del fascículo con células inflamatorias que invaden las fibras musculares (Figura 3) (14).
 Figura 3: En el panel de la izquierda y en derecha se ven biopsias en PM. Hay un intenso infiltrado mononuclear intersticial con alguna degeneración de miocitos.
Figura 3: En el panel de la izquierda y en derecha se ven biopsias en PM. Hay un intenso infiltrado mononuclear intersticial con alguna degeneración de miocitos.(Fuente UpToDate)
· En contraste a la DM hay fibras musculares anormales a lo largo de todo el fascículo, y no limitadas a una porción del mismo. No hay signos de vasculopatía ni deposición de complejos inmunes. Hay, sin embargo, evidencias de mecanismos inmunes mediados por células, con aumento de células T CD8+, que parecen reconocer un antígeno en la superficie de la fibra muscular (13), y hay una exagerada expresión del complejo mayor de histocompatibilidad clase I (MHC clase I) por las fibras musculares (17)
Las citoquinas proinflamatorias pueden contribuir a la debilidad muscular en ausencia de cambios inflamatorios histológicos evidentes. Tanto la interleukina 1 (IL-1) como el factor de necrosis tumoral alfa (TNF-alfa) están aumentados en el tejido muscular de los pacientes con DM-PM. Ambas citoquinas, así como la “up-regulation” deL complejo mayor de histocompatibilidad clase I(MHC clase I) en los miocitos pueden contribuir a los disturbios de la función muscular.(18)
Resumiendo, en la DM la lesión sería inicialmente por inmunocomplejos depositados en los vasos de la región perifascicular, con participación del complemento, que dispara un reclutamiento de células CD4 que producen lesión en zonas específicas del músculo. En cambio en la PM, la lesión es dentro mismo del fascículo muscular, y no perifascicular, participando células T CD8+, que parecen reconocer un antígeno en la superficie de la fibra muscular, y hay una exagerada expresión del complejo mayor de histocompatibilidad clase I (MHC clase I) por las fibras musculares, lo que atraería a los CD8 que finalmente producen la inflamación y el daño.
Manifestaciones clínicas
Las citoquinas proinflamatorias pueden contribuir a la debilidad muscular en ausencia de cambios inflamatorios histológicos evidentes. Tanto la interleukina 1 (IL-1) como el factor de necrosis tumoral alfa (TNF-alfa) están aumentados en el tejido muscular de los pacientes con DM-PM. Ambas citoquinas, así como la “up-regulation” deL complejo mayor de histocompatibilidad clase I(MHC clase I) en los miocitos pueden contribuir a los disturbios de la función muscular.(18)
Resumiendo, en la DM la lesión sería inicialmente por inmunocomplejos depositados en los vasos de la región perifascicular, con participación del complemento, que dispara un reclutamiento de células CD4 que producen lesión en zonas específicas del músculo. En cambio en la PM, la lesión es dentro mismo del fascículo muscular, y no perifascicular, participando células T CD8+, que parecen reconocer un antígeno en la superficie de la fibra muscular, y hay una exagerada expresión del complejo mayor de histocompatibilidad clase I (MHC clase I) por las fibras musculares, lo que atraería a los CD8 que finalmente producen la inflamación y el daño.
Manifestaciones clínicas
La DM y la PM son trastornos multisistémicos con una amplia variedad de potenciales hallazgos clínicos.
Debilidad muscular:
Debilidad muscular:
La debilidad muscular es la forma de presentación más común tanto en DM como en PM. El inicio es usualmente insidioso, con empeoramiento gradual en un período de varios meses antes de que el paciente busque atención médica. Sin embargo, ha sido descripto un inicio agudo.
La distribución de la debilidad es típicamente simétrica y proximal. La debilidad distal, si está presente, tiende a ser leve y no ocasiona trastornos funcionales significativos.
Las mialgias y el dolor a la palpación de las masas musculares ocurre en 25 a 50% de los casos. No es tan dolorosa como la polimialgia reumática, como la fibromialgia, ni como las miositis viral o bacteriana.
La atrofia muscular no se ve en estadios tempranos, pero puede ser vista en etapas tardías.
Hallazgos en piel:
La distribución de la debilidad es típicamente simétrica y proximal. La debilidad distal, si está presente, tiende a ser leve y no ocasiona trastornos funcionales significativos.
Las mialgias y el dolor a la palpación de las masas musculares ocurre en 25 a 50% de los casos. No es tan dolorosa como la polimialgia reumática, como la fibromialgia, ni como las miositis viral o bacteriana.
La atrofia muscular no se ve en estadios tempranos, pero puede ser vista en etapas tardías.
Hallazgos en piel:
Hay distintos tipos de rash, y están generalmente presentes al momento de la presentación, se ve en DM y no en PM:
· Signo de Gottron es un rash eritemato-escamoso simétrico sobre las articulaciones metacarpofalángicas y las interfalángicas. (Figura 4)

Figura 4: Una erupción escamosa sobre la superficie extensora de las metacarpofalánficas y los dedos en un paciente con DM. La lesión es llamada signo de Gottron y puede mimetizar la psoriasis
· Signo de Gottron es un rash eritemato-escamoso simétrico sobre las articulaciones metacarpofalángicas y las interfalángicas. (Figura 4)

Figura 4: Una erupción escamosa sobre la superficie extensora de las metacarpofalánficas y los dedos en un paciente con DM. La lesión es llamada signo de Gottron y puede mimetizar la psoriasis
Lesiones similares pueden se vistas en las caras extensoras de los codos y rodillas mimetizando la psoriasis.
· Rashen heliotropo, que es una erupción violácea en los párpados superiores, a menudo acompañado por inflamación del párpado (Figura 5).

Figura 5: Una erupción rojo purpúrea en el párpado superior (rash en heliotropo) acompañado de inflamación del párpado en paciente con DM. Es el rash más específico de la DM aunque solo lo presenta una minoría de los pacientes.
(Fuente UpToDte)
· Rashen heliotropo, que es una erupción violácea en los párpados superiores, a menudo acompañado por inflamación del párpado (Figura 5).

Figura 5: Una erupción rojo purpúrea en el párpado superior (rash en heliotropo) acompañado de inflamación del párpado en paciente con DM. Es el rash más específico de la DM aunque solo lo presenta una minoría de los pacientes.
(Fuente UpToDte)
· El signo del chal o de la manta, es un eritema difuso que ocurre en la parte superior del tórax y los hombros con forma de V sobre el cuello y tórax. (Figura 6). Es habitualmente fotosensible, y puede ser exacerbado por la luz ultravioleta.

Figura 6: Signo del chal o de la V en la DM.
· La eritrodermia se refiere a un eritema que recuerda al signo del chal pero que es visto en una variedad de áreas, incluyendo la región malar (Figura 7), y en la frente (Figura 8).

Figura 7: Eritrodermia facial en un paciente con DM

· Anormalidades periungueales, los capilares de los lechos enla DM pueden ser eritematosos y mostrar cambios vasculares similares a aquellos observados en otras enfermedades del tejido conectivo (por ej esclerodermia y lupus). Los capilares de los lechos ungueales pueden verse a simple vista, con áreas de dilatación y áreas mas finas, acompañado de eritema periungueal (Figura 9)

Figura 9: Eritema periungueal en DM

Figura 9: Eritema periungueal en DM
· Las manos mecánicas, que es una mano áspera y con crujidos de la piel en los extremos y bordes de los dedos, donde aparecen líneas sucias que recuerdan la de las personas que realizan trabajos duros con las manos. Esta lesión está clásicamente asociada con el síndrome anti-sintetasa, pero ha sido reportada con otros anticuerpos. (ver síndrome anti-sintetas más abajo). Figura 10

Figura 10: Manos mecánicas en paciente con PM y síndrome anti-sintetasa
· Cambios psoriasiformes deel cuero cabelludo. Estos recuerdan a la psoriasis, y ocurren en alto porcentaje de los pacientes con dermatomiositis. (Figura 11)


11: Cambios psoriasiformes en cuero cabelludo en paciente con DM.
· Eritema flagelado, que son estrías lineales en tronco (21) (Figura 12)

Figura 12: Eritema flagelado en DM.

Figura 12: Eritema flagelado en DM.
· Calcinosis cutis, que es la deposición de calcio dentro de la piel, ocurre comúnmente en la DM juvenil pero es inusual en los adultos.
Un número de otras más inusuales manifestaciones de piel incluyen la ictiosis, paniculitis, liquen plano, estrías atróficas blanco aporcelanadas, vesículas y bullas, malakoplakia, y mucinosis papular. (22)
Enfermedad pulmonar:
Un número de otras más inusuales manifestaciones de piel incluyen la ictiosis, paniculitis, liquen plano, estrías atróficas blanco aporcelanadas, vesículas y bullas, malakoplakia, y mucinosis papular. (22)
Enfermedad pulmonar:
La enfermedad pulmonar intersticial es una importante complicaciónde aproximadamente el 10% de los casos de DM y PM. Además, puede desarrollar fallo respiratorio, debido a compromiso del diafragma y debilidad de la pared torácica.
Cuando ocurre enfermedad intersticial ésta evoluciona a una insuficiencia respiratoria rápidamente progresiva, fallo respiratorio y muerte. Este compromiso intersticial ocurre en el contexto de anticuerpos anti-sintetasa y el síndrome anti-sintetasa.
Cáncer:
Un aumento de la tasa de cáncer ha sido descripto con mayor riesgo en DM que en PM (ver cáncer y dermato-polimiositis.
Enfermedad esofágica:
Enfermedad esofágica:
La debilidad del músculo estriado del tercio superior del esófago (y/o músculos orofaringeos) pueden conducir a disfagia, regurgitación nasal, y/o aspiración (23). El compromiso esofágico es más común en los ancianos y puede ocasionar neumonías aspirativas. (24)
Enfermedad cardíaca:
Enfermedad cardíaca:
El compromiso cardíaco con evidencia histológica de miocarditis está perfectamente descripto en DM y en PM (25). Sin embargo, el compromiso miocárdico suficientemente severo para causar fallo cardíaco es inusual.
Los pacientes con miositis pueden tener una fraccción CK-MB elevada, debido a expresión aumentada de la cadena CK-B del músculo esquelético inflamado (26), o a compromiso miocárdico por la miositis. Sin embargo, la razón más común para el aumento de los niveles de CK-MB es que la fracción MB está aumentada en el músculo en regeneración. En esos pacientes a veces se sospecha infarto de miocardio.
La troponina I cardíaca, un más específico y sensible marcador de daño cardíaco que la CK-MB, puede ser usada para establecer la causa de la elevación de la CK-MB en los pacientes con miositis. La troponina I en concentración normal asegura que no hay compromiso cardíaco.
Otros:
Los pacientes con miositis pueden tener una fraccción CK-MB elevada, debido a expresión aumentada de la cadena CK-B del músculo esquelético inflamado (26), o a compromiso miocárdico por la miositis. Sin embargo, la razón más común para el aumento de los niveles de CK-MB es que la fracción MB está aumentada en el músculo en regeneración. En esos pacientes a veces se sospecha infarto de miocardio.
La troponina I cardíaca, un más específico y sensible marcador de daño cardíaco que la CK-MB, puede ser usada para establecer la causa de la elevación de la CK-MB en los pacientes con miositis. La troponina I en concentración normal asegura que no hay compromiso cardíaco.
Otros:
Hay pacientes que tienen otras manifestaciones como fiebre, pérdida de peso, Raynaud, y poliartritis inflamatoria no erosiva.
Síndrome anti-sintetasa:
Síndrome anti-sintetasa:
Hasta el 30% de pacientes con DM o PM tienen una constelación de hallazgos llamados en conjunto “el síndrome anti-sintetasa”. Esos hallazgos incluyen un inicio relativamente agudo de la enfermedad, síntomas constitucionales (por ej fiebre), Raynaud, manos mecánicas, artritis, y enfermedad del intersticio pulmonar. Estos pacientes tienen anticuerpos anti-sintetasa, que tienen alta especificidad para DM y PM. (28,29)
En consideración con este síndrome han sido notados varios puntos:
En consideración con este síndrome han sido notados varios puntos:
· No todos los pacientes con anticuerpos anti-sintetas o aún los clasificados como de padecer síndrome anti-sintetasa tienen todas las manifestaciones de este síndrome.
· Este grupo de hallazgos clínicos o el cuadro clínico general no es específico para anticuerpos anti-sintetasa. Pacientes con otro tipo de anticuerpos, por ejemplo anti-PM-Scl, anti-U1RNP, pueden también presentarse con estos signos y síntomas.
· Finalmente, algunos pacientes con anticuerpos anti-sintetasa tienen relativamente poca miositis pero tienen otros hallazgos más prominentes de esta enfermedad como enfermedad del intersticio pulmonar.
DM amiopática:
· Este grupo de hallazgos clínicos o el cuadro clínico general no es específico para anticuerpos anti-sintetasa. Pacientes con otro tipo de anticuerpos, por ejemplo anti-PM-Scl, anti-U1RNP, pueden también presentarse con estos signos y síntomas.
· Finalmente, algunos pacientes con anticuerpos anti-sintetasa tienen relativamente poca miositis pero tienen otros hallazgos más prominentes de esta enfermedad como enfermedad del intersticio pulmonar.
DM amiopática:
La DM amiopática y la dermatomiositis sine miositis son términos usados para describir pacientes con el rash típico, y dermatopatología de DM quienes no tienen evidencias de miopatía. La mayoría de ellos finalmente en la evolución desarrollan miositis; sin embargo, el compromiso muscular puede aparecer tan tardiamente como más de 6 años después del inicio de la enfermedad (30-32)
Síndromes de superposición:
Síndromes de superposición:
DM y PM pueden superponerse con otras enfermedades del tejido conectivo, particularmente esclerodermia, lupus eritematoso sistémico, enfermedad mixta del tejido conectivo, artritis reumatoidea, y síndrome de Sjögren. La miopatía asociada a otra enfermedad del tejido conectivo varía desde clínicamente insignificante (con mínima elevación de enzimas musculares y mínimos cambios inflamatorios en la histología) hasta la típicamente severa PM o DM en la que la miopatía domina el cuadro clínico. (4,5,34)
Diagnóstico:
Diagnóstico:
El diagnóstico de DM o PM es sugerido por los hallazgos clínicos arriba mencionados. La historia médica puede ayudar a eliminar el uso de drogas prescriptas o ilícitas como causa del daño ( por ej colchicina, estatinas, hidroxicloroquina, alcohol, cocaína). Además, factores de riesgo para infección por HIV y elementos de enfermedades del tejido conectivo deben ser buscados.
Tanto las mediciones de los niveles de enzimas musculares como la investigación de la presencia de autoanticuerpos específicos juegan importantes roles en la evaluación de los pacientes con DM y PM.
Enzimas musculares:
Tanto las mediciones de los niveles de enzimas musculares como la investigación de la presencia de autoanticuerpos específicos juegan importantes roles en la evaluación de los pacientes con DM y PM.
Enzimas musculares:
La CPK, LDH, Aldolasa, aspartato aminotransferasa (AST), y alaninoaminotransferasa (ALT) son las enzimas musculares rutinariamente medidas en la evaluación de una miopatía. La sangre no debe extraerse después de la biopsia muscular, debido al trauma por la inserción de la aguja que puede conducir a elevación de las enzimas musculares.
En algún momento de la enfermedad, casi todos los pacientes con DM o PM tienen un aumento de al menos una enzima muscular; la mayoría tienen aumento de todas ellas.
En casos severos, la concentración de CPK puede estar elevada 50 veces. Aunque una correlación entre la severidad de la enfermedad y el nivel de elevación de las enzimas musculares puede ser visto (5) el grado de disfunción muscular puede ser mucho mayor que el sugerido por las enzimas.Un importante concepto es que el tratamiento de la DM y la PM debe ser guiado principalmente por la fuerza del paciente, y no por el nivel de enzimas musculares.
La ocurrencia de debilidad muscular con enzimas relativamente normales es más probable que ocurra en DM que en PM. Niveles de enzimas persistentemente bajos en un contexto de debilidad muscular obvia ocurre en pacientes con enfermedad avanzada y significativa pérdida de masa muscular.
Autoanticuerpos:
Los FAN son detectados por métodos standard de inmunofluorescencia, y están presentes hasta en un 80% de los pacientes con DM oPM (35,36). El testeo detallado de autoanticuerpos diseñados para identificar los autoanticuerpos responsables del patrón de FAN es mucho más útil. La investigación de autoanticuerpos específicos en el contexto de miopatías inflamatorias es útil en dos contextos:
En algún momento de la enfermedad, casi todos los pacientes con DM o PM tienen un aumento de al menos una enzima muscular; la mayoría tienen aumento de todas ellas.
En casos severos, la concentración de CPK puede estar elevada 50 veces. Aunque una correlación entre la severidad de la enfermedad y el nivel de elevación de las enzimas musculares puede ser visto (5) el grado de disfunción muscular puede ser mucho mayor que el sugerido por las enzimas.Un importante concepto es que el tratamiento de la DM y la PM debe ser guiado principalmente por la fuerza del paciente, y no por el nivel de enzimas musculares.
La ocurrencia de debilidad muscular con enzimas relativamente normales es más probable que ocurra en DM que en PM. Niveles de enzimas persistentemente bajos en un contexto de debilidad muscular obvia ocurre en pacientes con enfermedad avanzada y significativa pérdida de masa muscular.
Autoanticuerpos:
Los FAN son detectados por métodos standard de inmunofluorescencia, y están presentes hasta en un 80% de los pacientes con DM oPM (35,36). El testeo detallado de autoanticuerpos diseñados para identificar los autoanticuerpos responsables del patrón de FAN es mucho más útil. La investigación de autoanticuerpos específicos en el contexto de miopatías inflamatorias es útil en dos contextos:
· Diagnóstico de enfermedades del tejido conectivo asociada con miositis.
· Detección de autoanticuerpos específicos de miositis, que pueden tener mucha importancia al momento de proveer información diagnóstica, pronóstica y potencial compromiso de determinados órganos en el futuro.
Autoanticuerpos y enfermedades del tejido conectivo
Autoanticuerpos y enfermedades del tejido conectivo
La detección de anticuerpos anti-Ro, anti-La, anti-Sm, o anti-ribonucleoproteína (RNP) sugiere fuertemente el diagnóstico de miositis asociada o superpuesta a otra enfermedad del tejido conectivo (35) Como ejemplo, los anticuerpos anti-RNP están asociados con la superposición de miositis con hallazgos ya sea de esclerodermia o lupus eritematoso sistémico, entidad conocida como enfermedad mixta del tejido conectivo (37).
El diagnóstico preciso de una enfermedad del tejido conectivo es crítica para el manejo del paciente debido a las implicancias pronósticas y terapéuticas
Autoanticuerpos específicos de miositis
El diagnóstico preciso de una enfermedad del tejido conectivo es crítica para el manejo del paciente debido a las implicancias pronósticas y terapéuticas
Autoanticuerpos específicos de miositis
Varias categorías de autoanticuerpos dirigidos contra sintetasas RNA citoplasmáticas, otras proteínas citoplasmáticas, ribonucleoproteínas, y ciertos antígenos nucleares llamados autoanticuerpos específicos de miositis. (28,38-40) Esos autoanticuerpos ocurren en aproximadamente 30% de los pacientes con DM y PM.
Aunque no se conoce el rol específico de estos autoanticuerpos específicos, cada vez se piensa más fuertemente que tienen un papel patogénico en DM y PM.
Hay tres categorías de autoanticuerpos específicos de miositis: anticuerpos contra la aminoacil-tRNA sintetasa (anticuerpos anti-sintetasa), incluyendo anti-Jo-1; anticuerpos contra una partícula de reconocimiento de señal (anticuerpos anti-SRP); y anticuerpos anti-Mi-2, una helicasa nuclear.
Aunque no se conoce el rol específico de estos autoanticuerpos específicos, cada vez se piensa más fuertemente que tienen un papel patogénico en DM y PM.
Hay tres categorías de autoanticuerpos específicos de miositis: anticuerpos contra la aminoacil-tRNA sintetasa (anticuerpos anti-sintetasa), incluyendo anti-Jo-1; anticuerpos contra una partícula de reconocimiento de señal (anticuerpos anti-SRP); y anticuerpos anti-Mi-2, una helicasa nuclear.
· Anti-Jo-1, son los autoanticuerpos más específicos de miositis. Están dirigidos contra la histidil-tRNA sintetasa. Están fuertemente asociados a enfermedad del intersticio pulmonar, con el fenómeno de Raynaud, artritis, y manos mecánicas. (38,39)
· Anti-SRP, son anticuerpos contra la partícula de reconocimiento de señal (SRP) Se asocian a severa miopatía y enfermedad agresiva, dificultosa de controlar, aún con dosis altas de corticoides asociadas a inmunosupresores.
· Anti-Mi-2, son anticuerpos dirigidos contra la helicas afectada a la activación transcripcional. (42) Se asocian a inicio relativamente agudo de la DM clásica con eritrodermia y el signo del chal (37,38).
Autoanticuerpos específicos de miositis e histopatología
Hay variaciones variaciones histopatológicas de acuerdo al tipo de autoanticuerpo específico de miositis (44,49). Como ejemplos:
· El patrón histopatológico de anticuerpo anti-Jo-1 parece diferir del de los pacientes con anti-SRP positivos.
· El patrón histopatológico de los pacientes con el fenotipo de DM y anti-sintetasa o anti-SRP difiere del patrón histopatológico que no tienen autoanticuerpos. Por ejemplo, los pacientes con anticuerpos anti-Jo-1 positivos tienen atrofia perifascucular típica de la DM pero no la característica patología capilar. En contraste, los pacientes con anti-SRP tienen patología capilar pero no atrofia perifascicular.
Electromiografía y biopsia tisular:
· El patrón histopatológico de los pacientes con el fenotipo de DM y anti-sintetasa o anti-SRP difiere del patrón histopatológico que no tienen autoanticuerpos. Por ejemplo, los pacientes con anticuerpos anti-Jo-1 positivos tienen atrofia perifascucular típica de la DM pero no la característica patología capilar. En contraste, los pacientes con anti-SRP tienen patología capilar pero no atrofia perifascicular.
Electromiografía y biopsia tisular:
Además de los tests de laboratorio para enzimas musculares y autoanticuerpos, la electromiografía y la biopsia tisular de piel y/o músculo son importantes facetas de la evaluación de los pacientes con posible DM o PM.
Electromiografía (EMG):
Electromiografía (EMG):
El EMG muestra evidencias de aumentada irritabilidad en forma de una triada clásica:
· Actividad insercional aumentada y fibrilaciones espontáneas.
· Potenciales anormales miopáticos de baja amplitud, corta duración y polifásicos.
· Descargas repetitivas complejas.
· Actividad insercional aumentada y fibrilaciones espontáneas.
· Potenciales anormales miopáticos de baja amplitud, corta duración y polifásicos.
· Descargas repetitivas complejas.
Además, en la miopatía temprana, hay reclutamiento temprano.
Un patrón normal de EMG es inusual en pacientes con DM oPM.
El EMG puede ser compatible con DM oPM pero no es diagnóstico. Hallazgos similares ocurren en varias miopatías infecciosas, tóxicas, o metabólicas.
Sin embargo, el EMG es de gran valor en diferenciar la debilidad de origen miopático, de la de origen neuropático, tal como la ELA (esclerosis lateral amiotrófica), polineuropatía periférica, o mistenia gravis.
El EMG es útil para elegir el sitio de la biopsia muscular
Biopsia de piel en DM
Un patrón normal de EMG es inusual en pacientes con DM oPM.
El EMG puede ser compatible con DM oPM pero no es diagnóstico. Hallazgos similares ocurren en varias miopatías infecciosas, tóxicas, o metabólicas.
Sin embargo, el EMG es de gran valor en diferenciar la debilidad de origen miopático, de la de origen neuropático, tal como la ELA (esclerosis lateral amiotrófica), polineuropatía periférica, o mistenia gravis.
El EMG es útil para elegir el sitio de la biopsia muscular
Biopsia de piel en DM
La confirmación histopatológica del diagnóstico de DM debe ser lograda cada vez que sea posible. La biopsia de cualquier manifestación de la enfermedad en piel, incluyendo el signo de Gottron, el signo del chal, y la eritrodermia puede proveer confirmación del diagnóstico en un contexto clínico apropiado.
Así, la biopsia de piel puede sellar el diagnóstico y evitar la necesidad de biopsia muscular en un paciente con debilidad en un patrón típico de miopatía inflamatoria (simétrica, predominio del compromiso proximal sobre el distal), elevación de enzimas musculares y hallazgos cutáneos característicos de DM.
A la microscopía de luz, las lesiones de la DM usualmente demuestran leve atrofia de la epidermis con cambios vacuolares en la capa basal. Un infiltrado linfoide perivascular a menudo prevalece en la dermis. (31)
La observación con microscopía de luz es esencial para hacer el diagnóstico histológico de DM. La inmunofluorescencia revela una dermatitis de interfase (deposición de proteínas del complemento e inmunoglobulina en la unión dermoepidérmica) que generalmente es indistinguible en la microscopía de luz a la del lupus eritematoso sistémico (Histologia 4)
Biopsia muscular.
Así, la biopsia de piel puede sellar el diagnóstico y evitar la necesidad de biopsia muscular en un paciente con debilidad en un patrón típico de miopatía inflamatoria (simétrica, predominio del compromiso proximal sobre el distal), elevación de enzimas musculares y hallazgos cutáneos característicos de DM.
A la microscopía de luz, las lesiones de la DM usualmente demuestran leve atrofia de la epidermis con cambios vacuolares en la capa basal. Un infiltrado linfoide perivascular a menudo prevalece en la dermis. (31)
La observación con microscopía de luz es esencial para hacer el diagnóstico histológico de DM. La inmunofluorescencia revela una dermatitis de interfase (deposición de proteínas del complemento e inmunoglobulina en la unión dermoepidérmica) que generalmente es indistinguible en la microscopía de luz a la del lupus eritematoso sistémico (Histologia 4)
Biopsia muscular.
Para la mayoría de los casos de miositis, el test definitivo para establecer el diagnóstico de miopatía y excluir otras causas de debilidad muscular, es la biopsia. La biopsia debe ser obtenida de un músculo que tiene debilidad a la exploración. El músculo usualmente elgido es el cuádriceps o el deltoides. La biopsia contralateral de un músculo que mostró alteración en el EMG suele dar el diagnóstico.
Los músculos con severa debilidad, marcada atrofia, o recientes tests EMG deben ser evitados. Además, la biopsia de los músculos de la pantorrilla son desaconsejados. Si el examen físico o la EMG fallan en identificar un músculo apropiado para la biopsia, la RMN puede revelar aumento de la señal en T2 en músculos que son aptos para ser biopsiados (por ej deltoides, bíceps, cuádriceps)
Una biopsia abierta es preferida a una biopsia con aguja debido a que se puede obtener mayor muestra de tejido y la orientación de las fibras puede observarse y preservarse mejor.
Un adecuado procesamiento de la biopsia muscular es esencial. Una vez obtenida la muestra, esta debe ser apropiadamente preservada para el procesamiento posterior. Una parte del tejido debe ser fijado de rutina, para microscopía óptica y electrónica, y otra parte debe ser congelada en isopentano enfriado con nitrógeno líquido para estudios bioquímicos.
Resonancia magética (RMN)
Los músculos con severa debilidad, marcada atrofia, o recientes tests EMG deben ser evitados. Además, la biopsia de los músculos de la pantorrilla son desaconsejados. Si el examen físico o la EMG fallan en identificar un músculo apropiado para la biopsia, la RMN puede revelar aumento de la señal en T2 en músculos que son aptos para ser biopsiados (por ej deltoides, bíceps, cuádriceps)
Una biopsia abierta es preferida a una biopsia con aguja debido a que se puede obtener mayor muestra de tejido y la orientación de las fibras puede observarse y preservarse mejor.
Un adecuado procesamiento de la biopsia muscular es esencial. Una vez obtenida la muestra, esta debe ser apropiadamente preservada para el procesamiento posterior. Una parte del tejido debe ser fijado de rutina, para microscopía óptica y electrónica, y otra parte debe ser congelada en isopentano enfriado con nitrógeno líquido para estudios bioquímicos.
Resonancia magética (RMN)
La RMN ha emergido como una importante técnica en la evaluación clínica de los pacentes con DM y PM (53). Las imágenes por RMN pueden demostrar áreas de inflamación muscular, edema con miositis activa, fibrosis, y calcificación. A diferencia de la biopsia muscular, la RMN puede explorar grandes áreas de músculo, evitando así errores en la toma de muestras. Dado que la RMN es no invasiva, tiene una ventaja adicional de permitir el seguimiento seriado, quepuede ser útil en evaluar la respuesta terapéutica.
Algunos centros la utilizan de rutina en cualquier miopatía inflamatoria. Sin embargo, estos estudios no reemplazan la biopsia ni la EMG en la realización del diagnóstico.
Otra modalidad es la espectroscopía por resonancia, que provee información del metabolismo muscular comparando la relación del fósforo muscular contenido en la fosfocreatina y el nivel de fósforo inorgánico. Esta relación está disminuída en el músculo anormal en el cual la generación de compuestos de alta energía tal como la fosfocreatina está reducida. Es un método muy sensible para detectar anormalidades en la DM amiopática, donde la fuerza muscular y las enzimas son normales.
Diagnóstico diferencial:
Algunos centros la utilizan de rutina en cualquier miopatía inflamatoria. Sin embargo, estos estudios no reemplazan la biopsia ni la EMG en la realización del diagnóstico.
Otra modalidad es la espectroscopía por resonancia, que provee información del metabolismo muscular comparando la relación del fósforo muscular contenido en la fosfocreatina y el nivel de fósforo inorgánico. Esta relación está disminuída en el músculo anormal en el cual la generación de compuestos de alta energía tal como la fosfocreatina está reducida. Es un método muy sensible para detectar anormalidades en la DM amiopática, donde la fuerza muscular y las enzimas son normales.
Diagnóstico diferencial:
La DM y PM deben ser distinguidos de otras condiciones que causan debilidad muscular, con o sin elevación de enzimas musculares. El diagnóstico diferencial incluye otras miopatías inflamatorias, enfermedades de motoneurona, miastenia gravis, y las distrofias musculares. Además, una variedad de trastornos heredados, metabólicos, inducidos por drogas, endocrinos y miopatías infecciosas deben ser consideradas (55)
Miositis por cuerpos de inclusión (MCI)
Miositis por cuerpos de inclusión (MCI)
Es la miopatía inflamatoria más comúnmente confundida con la PM. Muchos “casos refractarios” de PM son en realidad MCI. Establecer el diagnóstico diferencial entre estas dos entidades es fundamental, ya que el pronóstico es completamente diferente.
En contraste con la PM, la MCI es más insidiosa en su inicio y con debilidad distal más prominente. Se da más frecuentemente en los hombres al revés que la DM y la PM, y se ve generalmente en edades más avanzadas de la vida que la DM y la PM. La atrofia muscular y debilidad de las muñecas y los flexores de los dedos en las extremidades superiores y los cuádriceps y músculos tibiales anteriores en las piernas es característico
En promedio, el nivel de enzimas musculares es menor que en PM, aunque pueden existir elevaciones importantes. La presencia de cuerpos de inclusión en la biopsia es característica de esta entidad, pero la obtención de una única biopsia da un rédito diagnóstico de sólo 20 a 30% (56).
La RMN puede ayudar a distinguir entre PM y MCI. Mientras que, cambios sugestivos de inflamación son notados a lo largo de los planos fasciales en PM, tales cambios son observados a lo largo de todo el músculo en la MCI. La infiltración grasa y la atrofia son más prominentes en la MCI que en la PM.
Hipotiroidismo:
En contraste con la PM, la MCI es más insidiosa en su inicio y con debilidad distal más prominente. Se da más frecuentemente en los hombres al revés que la DM y la PM, y se ve generalmente en edades más avanzadas de la vida que la DM y la PM. La atrofia muscular y debilidad de las muñecas y los flexores de los dedos en las extremidades superiores y los cuádriceps y músculos tibiales anteriores en las piernas es característico
En promedio, el nivel de enzimas musculares es menor que en PM, aunque pueden existir elevaciones importantes. La presencia de cuerpos de inclusión en la biopsia es característica de esta entidad, pero la obtención de una única biopsia da un rédito diagnóstico de sólo 20 a 30% (56).
La RMN puede ayudar a distinguir entre PM y MCI. Mientras que, cambios sugestivos de inflamación son notados a lo largo de los planos fasciales en PM, tales cambios son observados a lo largo de todo el músculo en la MCI. La infiltración grasa y la atrofia son más prominentes en la MCI que en la PM.
Hipotiroidismo:
La miopatía asociada al hipotiroidismo puede mimetizar la presentación de una miopatía inflamatoria, causando debilidad muscular y elevación de enzimas musculares
Miopatía inducida por drogas:
Miopatía inducida por drogas:
Un número de drogas puede producir miopatía inflamatorias. Ellas incluyen corticosteroides, estatinas, antipalúdicos, colchicina, penicilamina, alcohol, cocaína, y ciertas drogas antiretrovirales.
Infección por HIV:
Infección por HIV:
La debilidad en un paciente infectado por HIV representa un desafío diagnóstico. Además de causar la debilidad propia de toda enfermedad crónica y caquexia, la infección por HIV puede estar asociada a miopatía inflamatoria que es idéntica a la PM. La miopatía puede presentarse ya sea como manifestación de infección por HIV u ocurrir tardiamente en el curso de la infección. También algunas drogas antiretrovirales pueden causar miopatía.
Esclerosis lateral amiotrófica (ELA)
Esclerosis lateral amiotrófica (ELA)
ELA, también llamada enfermedad de neurona motora inferior, tiene un número de elementos clínicos que la diferencian de la DM y de la PM
· La ELA puede presentarse con compromiso distal más que proximal y en su inicio no es generalmente simétrica.
· Signos de fibras largas como liberación piramidal son típicos en la ELA ya que la disfunción es tanto de neurona motora inferior como superior.
· La ELA no está asociada con cambios miopáticos en el EMG debido a que el problema es la muerte neuronal.
· Las enzimas musculares usualmente son normales en ELA aunque puede haber elevaciones de CPK hasta aproximadamente 1000.
Miastenia gravis (MG):
· La ELA puede presentarse con compromiso distal más que proximal y en su inicio no es generalmente simétrica.
· Signos de fibras largas como liberación piramidal son típicos en la ELA ya que la disfunción es tanto de neurona motora inferior como superior.
· La ELA no está asociada con cambios miopáticos en el EMG debido a que el problema es la muerte neuronal.
· Las enzimas musculares usualmente son normales en ELA aunque puede haber elevaciones de CPK hasta aproximadamente 1000.
Miastenia gravis (MG):
MG es un trastorno de la unión neuromuscular, causado por anticuerpos contra el receptor de acetilcolina. Aunque el examen físico clásico en la MG es la fatigabilidad desencadenada por el ejercicio, ocasionalmente puede haber debilidad difusa sin fatigabilidad al ejercicio.
MG es distinguida de la PM por la frecuente afectación de la musculatura facial, enzimas musculares normales, cambios característicos en la EMG y la presencia de anticuerpos contra el receptor de acetilcolina.
En contraste a la MG, la DM y la PM raramente comprometen los músculos oculomotores. Una condición relacionada con MG es el síndroe de Eaton-Lambert, que puede mimetizar la PM y la DM dado que no afecta los músculos extraoculares.
Distrofias musculares y distrofia miotónica:
MG es distinguida de la PM por la frecuente afectación de la musculatura facial, enzimas musculares normales, cambios característicos en la EMG y la presencia de anticuerpos contra el receptor de acetilcolina.
En contraste a la MG, la DM y la PM raramente comprometen los músculos oculomotores. Una condición relacionada con MG es el síndroe de Eaton-Lambert, que puede mimetizar la PM y la DM dado que no afecta los músculos extraoculares.
Distrofias musculares y distrofia miotónica:
Las distrofias musculares son un grupo heredado de trastornos miopáticos progresivos que resultan de un defecto en un número de genes necesarios para una función muscular normal. Los pacientes con distrofia muscular ocasionalmente tienen un infiltrado inflamatorio en endomisio. Esto puede causar confusión, particularmente en la distrofia facio-escápulo-humeral, y en la distrofia de cintura pelviana. Sin embargo, el infiltrado inflamatorio de las distrofias típicamente está limitado a áreas adyacentes a las fibras necróticas.
La miopatía miotónica proximal, también conocida como distrofia miotónica tipo 2 debe ser también considerada en el diagnóstico diferencial.
Miopatías metabólicas heredadas:
La miopatía miotónica proximal, también conocida como distrofia miotónica tipo 2 debe ser también considerada en el diagnóstico diferencial.
Miopatías metabólicas heredadas:
Las miopatías metabólicas heredadas tales como los trastornos en el metabolismo de los carbohidratos y de los lípidos como las deficiencias de carnitina y mioadenilato deaminasa. Estas se caracterizan por episodios intermitentes de dolor muscular agudo usualmente inducidos por ejercicio. Esos episodios se acompañan de mioglobinuria. Ocasionalmente los pacientes desarrollan debilidad crónica después de años de episodios repetidos. (58)
Otros: Una variedad de miopatías puede mimetizar a la PM.
Las infecciones virales o bacterianas (piomiositis), la inmovilización, y el trauma, pueden caracterizarse por presentación aguda, fulminante, a menudo acompañado de rabdomiolisis. Sin embargo, la rabdomiolisis ha sido descripta en casos de miopatías inflamatorias (59,60).
La debilidad muscular ocurriendo en múltiples miembros de la familia no siempre es debido a defectos metabólicos heredados o distrofias, pero puede resultar del desarrollo de miositis idiopática en varios miembros de la misma familia.
Los hallazgos clínicos de la miopatía inflamatoria idiopática familiar son similares a aquellos con enfermedad esporádica, aunque la frecuencia de autoanticuerpos específicos de miositis es menor en la forma familiar. (61)
Una enfermedad que es clínica e histopatológicamente similar a la PM es la descripta en la enfermedad de transplante versus huésped. La frecuencia estimada es 0,6%. El inicio de la miositis es típicamente 1 año después del transplante (63)
Algunos raros pacientes se presentan con miositis focal, que, usualmente, pero no siempre progresa a la generalizada típica con el tiempo. (19)
La miopatía amiloide, que puede ocurrir en amiloidosis relacionada a inmunoglobulinas o amiloidosis familiar. (64)
Miopatía sarcoide.
Infecciones parasitarias por ejemplo triquinelosis.
Otros: Una variedad de miopatías puede mimetizar a la PM.
Las infecciones virales o bacterianas (piomiositis), la inmovilización, y el trauma, pueden caracterizarse por presentación aguda, fulminante, a menudo acompañado de rabdomiolisis. Sin embargo, la rabdomiolisis ha sido descripta en casos de miopatías inflamatorias (59,60).
La debilidad muscular ocurriendo en múltiples miembros de la familia no siempre es debido a defectos metabólicos heredados o distrofias, pero puede resultar del desarrollo de miositis idiopática en varios miembros de la misma familia.
Los hallazgos clínicos de la miopatía inflamatoria idiopática familiar son similares a aquellos con enfermedad esporádica, aunque la frecuencia de autoanticuerpos específicos de miositis es menor en la forma familiar. (61)
Una enfermedad que es clínica e histopatológicamente similar a la PM es la descripta en la enfermedad de transplante versus huésped. La frecuencia estimada es 0,6%. El inicio de la miositis es típicamente 1 año después del transplante (63)
Algunos raros pacientes se presentan con miositis focal, que, usualmente, pero no siempre progresa a la generalizada típica con el tiempo. (19)
La miopatía amiloide, que puede ocurrir en amiloidosis relacionada a inmunoglobulinas o amiloidosis familiar. (64)
Miopatía sarcoide.
Infecciones parasitarias por ejemplo triquinelosis.
Tratamiento inicial de la DM y PM en adultos.
Predictores de resultados:
La severidad de la enfermedad en DM y PM es altamente variable, yendo desde debilidad leve que responde bien al tratamiento, a la disfunción muscular generalizada y que no responde a ninguna modalidad de tratamiento. Algunos elementos clínicos y de laboratorio están asociados a peor pronóstico.
Predictores de pronóstico:
Predictores de pronóstico:
La mayoría de los estudios que han investigado pronóstico no distinguieron entre DM y PM. Había seguramente entre estos casos, muchas miositis por cuerpos de inclusión, que tienen peor pronóstico, y que no fueron descriptos hasta 1980.
Los siguientes elementos estuvieron relacionados a peor pronóstico:
1) Retraso en la iniciación del tratamiento por más de 6 meses del comienzo de los síntomas.
1) Retraso en la iniciación del tratamiento por más de 6 meses del comienzo de los síntomas.
2) Mayor debilidad en la presentación.
3) Presencia de disfagia.
4) Debilidad de los músculos respiratorios.
5) Enfermedad pulmonar intersticial.
6) Cáncer asociado.
7) Compromiso cardiovascular.
Tipos de miositis:
Un nuevo approach a la clasificación de miopatías inflamatorias sugiere que los pacientes con PM tienen menos probabilidad de responder a los glucocorticoides solos, que los pacientes con DM, o con miositis asociadas a enfermedades del tejido conectivo (las llamadas miositis de superposición).
· Entre los pacientes con PM, 50% fueron refractarios a los glucocorticoides.
· Entre los pacientes con DM, 87% respondieron a los cursos iniciales de glucocorticoides. Sin embargo, 92% presentaron recidivas cuando se descendió la dosis de corticosteroides.
· Entre los pacientes con miositis de superposición, la tasa de respuesta a los glucocorticoides varió desde 89 a 100%
Ciertos autoanticuerpos específicos de miositis pueden definir clínicamente distintos subgrupos que tienen algún valor predictivo de la respuesta al tratamiento.
El autoanticuerpo específico de miositis más común en DM y en PM es el anticuerpo anti-jo-1. Los pacientes con anti-jo-1, tienen respuestas incompletas al tratamiento, y peor pronóstico a largo plazo. El peor escenario es la evolución a compromiso intersticial pulmonar.
Algunos pacientes con anticuerpos contra partícula reconocimiento de señal ( anti SRP) (anti-signal recognition particle), tienen un más fulminante inicio de debilidad proximal, muy altos niveles séricos de CPK, y biopsias musculares que muestran necrosis y regeneración de fibras musculares, pero poca inflamación. El tratamiento temprano con corticosteroides es beneficioso en algunos de estos pacientes.
Los pacientes con anti-Mi-2, que solo ocurre en DM, a menudo tienen un inicio fulminante, y hallazgos cutáneos floridos. Sin embargo, responden bien al tratamiento y tienen buen pronóstico a largo plazo.
Resumiendo, en el futuro, los anticuerpos específicos de miositis jugarán un rol importante en la selección del tratamiento para DM y PM y en establecer un pronóstico.
Ciertos autoanticuerpos específicos de miositis pueden definir clínicamente distintos subgrupos que tienen algún valor predictivo de la respuesta al tratamiento.
El autoanticuerpo específico de miositis más común en DM y en PM es el anticuerpo anti-jo-1. Los pacientes con anti-jo-1, tienen respuestas incompletas al tratamiento, y peor pronóstico a largo plazo. El peor escenario es la evolución a compromiso intersticial pulmonar.
Algunos pacientes con anticuerpos contra partícula reconocimiento de señal ( anti SRP) (anti-signal recognition particle), tienen un más fulminante inicio de debilidad proximal, muy altos niveles séricos de CPK, y biopsias musculares que muestran necrosis y regeneración de fibras musculares, pero poca inflamación. El tratamiento temprano con corticosteroides es beneficioso en algunos de estos pacientes.
Los pacientes con anti-Mi-2, que solo ocurre en DM, a menudo tienen un inicio fulminante, y hallazgos cutáneos floridos. Sin embargo, responden bien al tratamiento y tienen buen pronóstico a largo plazo.
Resumiendo, en el futuro, los anticuerpos específicos de miositis jugarán un rol importante en la selección del tratamiento para DM y PM y en establecer un pronóstico.
Terapia inicial:
El objetivo del tratamiento es mejorar la fuerza muscular, y evitar el desarrollo de complicaciones extramusculares. En pacientes con DM, la resolución de la enfermedad cutánea es un objetivo adicional.
A pesar de ausencia de estudios controlados contra placebo, los corticosteroides son la piedra angular del tratamiento para DM y PM. Algunos clínicos también empiezan el tratamiento con agentes “ahorradores de corticosteroides”, generalmente azatioprina o metotrexato, particularmente en pacientes severamente enfermos.
Corticosteroides.
A pesar de ausencia de estudios controlados contra placebo, los corticosteroides son la piedra angular del tratamiento para DM y PM. Algunos clínicos también empiezan el tratamiento con agentes “ahorradores de corticosteroides”, generalmente azatioprina o metotrexato, particularmente en pacientes severamente enfermos.
Corticosteroides.
Los médicos deben informar a los pacientes al comenzar la terapia que tendrán que seguir tomando prednisona, o equivalentes en dosis que irán descendiéndose, al menos durante un año, dependiendo de la respuesta a la terapia y del control de la enfermedad. La información y discusión con el paciente de los efectos colaterales de los corticosteroides es esencial, para que ellos sepan a que anticiparse.
No hay un régimen corticoideo standard para el comienzo del tratamiento de las miopatías inflamatorias, pero se pueden aplicar 2 principios básicos para todas ellas:
No hay un régimen corticoideo standard para el comienzo del tratamiento de las miopatías inflamatorias, pero se pueden aplicar 2 principios básicos para todas ellas:
1) La iniciación del tratamiento con altas dosis por varios meses para establecer el control de la enfermedad.
2) Descenso lento hasta llegar a la menor dosis efectiva con una duración de entre 9 meses y 1 año.
Terapia inicial con corticosteroides.
Terapia inicial con corticosteroides.
La terapia con corticosteroides en DM y en PM es típicamente iniciada con prednisona o metilprednisona a la dosis de 1 mg/kg de peso por día, hasta una dosis máxima de 80 mg/día. Pulsos de metilprednisolona (1000 mg/día por 3 días) puede ser utilizada al comienzo de la terapia para pacientes severamente enfermos.
Para las primeras 4 a 6 semanas de terapia, la prednisona se continúa a 1 mg/kg de peso por día con evaluación periódica de las respuesta clínica. El mantenimiento de la prednisona a esa dosis más allá de 6 semanas no debe hacerse debido a que a partir de entonces existe el riesgo potencial de desarrollo de miopatía corticosteroidea.
Evaluación de la respuesta al tratamiento
La respuesta a los corticosteroides debe ser medida a las pocas semanas de comenzado el tratamiento. Algunos pacientes comienzan a mostrar mejoramiento en la fuerza muscular en algunas semanas. Otros, mejoran lentamente, sin mejoría evidente hasta 3 meses. En la DM, la respuesta cutánea al tratamiento se comporta igual que la miopatía, algunos responden rápidamente, y otros tardan varias semanas o meses.
Una vez comenzado el tratamiento, el clínico debe seleccionar varios grupos musculares para testear regularmente y evaluar la respuesta terapéutica.
Una vez comenzado el tratamiento, el clínico debe seleccionar varios grupos musculares para testear regularmente y evaluar la respuesta terapéutica.
Los siguientes grupos musculares son a menudo testeados:
1) Cuádriceps. Decir al paciente que cruce los brazos, y que trate de incorporarse (pararse) de la silla, usando sólo los músculos proximales de las piernas.
2) Deltoides: Decir al paciente que flexione el brazo a nivel del codo, y que después intente separar el brazo del tórax contra la oposición del médico examinador.
3) Flexores del cuello. Decir al paciente que empuje su cabeza hacia adelante mientras el examinador opone resistencia presionando sobre su frente.
El principio central de la respuesta al tratamiento es el mejoramiento de la fuerza muscular, que es el mejor indicador de respuesta clínica, mucho mayor que la concentración de enzimas musculares.
El descenso de las enzimas musculares séricas debe esperarse dentro de 2 semanas de la iniciación de la terapia, pero la normalización puede tomar un tiempo considerablemente mayor. Ajustar la dosis de prednisona en un intento de normalizar la concentración de enzimas musculares puede llevar a sobretratamiento.
Descenso de la dosis de corticosteroides.
El descenso de las enzimas musculares séricas debe esperarse dentro de 2 semanas de la iniciación de la terapia, pero la normalización puede tomar un tiempo considerablemente mayor. Ajustar la dosis de prednisona en un intento de normalizar la concentración de enzimas musculares puede llevar a sobretratamiento.
Descenso de la dosis de corticosteroides.
Después de 4 a 6 semanas se comienza el descenso de la dosis. Si el paciente no ha mostrado signos clínicos de mejoría hasta este momento, se debe agregar agentes “ahorradores de corticosteroides”, si es que no se habían comenzado junto con la prednisona desde el inicio.
No hay un régimen standard de descenso de dosis para las miopatías inflamatorias, pero una de las formas es la siguiente:
Asumiendo que se empezó con 60 mg/día de prednisona, y se mantuvo esta dosis 6 semanas, el descenso requerirá 26 semanas hasta llegar a la dosis de 5 mg:
No hay un régimen standard de descenso de dosis para las miopatías inflamatorias, pero una de las formas es la siguiente:
Asumiendo que se empezó con 60 mg/día de prednisona, y se mantuvo esta dosis 6 semanas, el descenso requerirá 26 semanas hasta llegar a la dosis de 5 mg:
· La prednisona debe ser descendida 10 mg por semana hasta llegar a 40 mg/día.
· Después de 1 semana de 40 mg por día, la prednisona debe ser descendida de a 5 mg por semana, hasta llegar a 20 mg por semana hasta llegar a 20 mg/día.
· Después de 1 semana tomando 20 mg, la dosis de prednisona debe ser descendida 2,5 mg por semana hasta alcanzar 10 mg por día.
· Después de 1 semana de tomar 10 mg por día, la prednisona debe ser descendida 1 mg por semana hasta llegar a 5 mg por día.
Cambios en este esquema deben realizarse según criterio médico, si la miopatía experimenta recidivas durante el descenso.
Descensos de la dosis de corticosteroides aún mayores (menos de 5 mg por día) puede considerarse si el paciente ha logrado y mantenido un buen control de la enfermedad. El retiro de la prednisona nunca debe ser más rápido de 1 mg cada 2 semanas en esta etapa del tratamiento.
El paciente debe ser monitoreado cuidadosamente durante el período de descenso, observándose y buscándose signos de recurrencia de la debilidad muscular, complicaciones extramusculares de la DM o de la PM, y buscando signos de toxicidad corticoidea. La recurrencia de debilidad proximal en un paciente tratado por períodos prolongados con corticosteroides, plantea el siguiente acertijo: es la debilidad muscular secundaria a la miopatía inflamatoria, o es producto de miopatía corticosteroidea?
Descenso a días alternos.
Aunque algunos clínicos usan el descenso de los corticoides a días alternos para el tratamiento de las miopatías inflamatorias, hay pocas evidencias de que este régimen tenga menor toxicidad comparado con la terapia convencional descripta arriba.
Respuesta a los corticoides
Respuesta a los corticoides
Más de 80% de los pacientes con miopatías inflamatorias, mejoran con corticosteroides solos. Sin embargo, como se dijo antes, tanto como el 50% de las PM no responden al tratamiento con corticosteroides sólos. Más aún, entre los pacientes que no responden, la mayoría no vuelve a recuperar totalmente su fuerza muscular.
Aparentes fallos de respuesta corticosteroidea.
En el contexto de un fallo aparente de respuesta a corticosteroides, antes de comenzar un aumento de la inmunosupresión tres posibles causas deben ser investigadas antes:
1) Considerar error de diagnóstico, y sospechar diagnósticos alternativos como miositis por cuerpos de inclusión, distrofia muscular, o hipotiroidismo. Repetir la biopsia muscular puede ser necesario.
2) Si el diagnóstico de DM o PM es confirmado, debe considerarse la miopatía por corticosteroides, sobre todo si el paciente permanece con debilidad permanente o recurrente, en el contexto de enzimas musculares normales. En estos casos, el EMG puede ser de ayuda. La persistencia de potenciales de fibrilación y ondas positivas agudas son sugestivas de miositis. Debido a que la naturaleza parcelar (“parcheada”) de la inflamación, y al hecho de que la inactividad puede causar atrofia de fibras musculares tipo2, la biopsia muscular no siempre ayuda en la distinción de miopatía inducida por corticosteroides y miopatía mal controlada. El rol de la RMN en estos casos todavía es incierto. Un ensayo empírico de descenso de la prednisona en estos casos observando la respuesta es un approach a este dilema.
3) Una enfermedad maligna no diagnosticada puede ser la causa de una miositis que no responde al tratamiento. La mayoría de los cánceres asociados a miositis se diagnostican dentro de los dos años desde el comienzo de la miositis.
Cuando se evalúa a un paciente con miositis en busca de alguna neoplasia asociada, el approach recomendado es una historia clínica detallada, y un examen físico minucioso, exámenes de laboratorio de rutina, y Rx de tórax. No se justifican la solicitud de estudios sin sentido, a no ser que de acuerdo a la edad del paciente correspondan por screening oncológico. Este tema será desarrollado más abajo.
Agentes “ahorradores de corticosteroides”.
Algunos clínicos inician de entrada el tratamiento con estos agentes asociados a los corticosteroides. Otros los reservan para los fracasos de los corticosteroides solos. Es aconsejable comenzar con estos agentes de entrada junto a los esteroides.
Actualmente, los agentes de primera línea son azatioprina (50 mg/día) o metotrexato (MTX) (15mg/semana). Esas drogas no han sido comparadas entre si en ensayos clínicos. Debido a la toxicidad hepática del MTX, se prefiere la azatioprina, sobre todo en pacientes con afectación pulmonar asociada a la miositis, enfermedad hepática de base, o alcoholismo.
Efectos adversos del MTX y azatioprina
Una reacción de tipo gripal con fiebre y síntomas gastrointestinales desarrollan en hasta 12% de los pacientes tratados con azatioprina, y requieren discontinuación de la droga. Otros efectos incluyen supresión de la médula ósea, pancreatitis, y toxicidad hepática. El uso a largo plazo aumenta el riesgo de cáncer.
Los recuentos hematológicos deben realizarse inicialmente 1 vez por mes y después cada 3 meses.
Con MTX, se pueden ver estomatitis, síntomas gastrointestinales, y leucopenia con dosis bajas. El riesgo puede ser minimizado con la administración de ácido fólico (1 a 2 mg/día) o en pacientes que no responden al ácido fólico usar ácido folínico (5 mg/semana, 8 a 12 horas después de la dosis de MTX).
Debido a su hepatotoxicidad el uso de MTX, el uso de esta droga es problemática entre los que toman alcohol, aunque sea en pequeñas cantidades.
No hay evidencias de que la toxicidad pulmonar con MTX sea más común en pacientes con enfermedad pulmonar asociada a la miositis.
Duración de la terapia
Los recuentos hematológicos deben realizarse inicialmente 1 vez por mes y después cada 3 meses.
Con MTX, se pueden ver estomatitis, síntomas gastrointestinales, y leucopenia con dosis bajas. El riesgo puede ser minimizado con la administración de ácido fólico (1 a 2 mg/día) o en pacientes que no responden al ácido fólico usar ácido folínico (5 mg/semana, 8 a 12 horas después de la dosis de MTX).
Debido a su hepatotoxicidad el uso de MTX, el uso de esta droga es problemática entre los que toman alcohol, aunque sea en pequeñas cantidades.
No hay evidencias de que la toxicidad pulmonar con MTX sea más común en pacientes con enfermedad pulmonar asociada a la miositis.
Duración de la terapia
La mayoría de los pacientes y los clínicos prefieren discontinuar antes los corticosteroides que azatioprina o MTX.
El descenso de la dosis de azatioprina y MTX debe ser realizada a intervalos mensuales después que el paciente entró en remisión, con la idea de suspenderlos a los 6 meses de comenzados.
Seguimiento a largo plazo.
El descenso de la dosis de azatioprina y MTX debe ser realizada a intervalos mensuales después que el paciente entró en remisión, con la idea de suspenderlos a los 6 meses de comenzados.
Seguimiento a largo plazo.
Los efectos colaterales de la terapia vistos en pacientes con miositis a largo plazo son:
· Aspecto cushingoide (71%).
· Síntomas psicológicos o psiquiátricos (35%).
· Osteoporosis (29%).
· Infecciones (29%)
En un seguimiento de 110 pacientes, 34 murieron en el curso del seguimiento, 18 por causas relacionadas con la miositis, 7 de cáncer, 4 de complicaciones pulmonares y 4 por complicaciones de la medicación.
Entre los 110 pacientes (84% de 131 sobrevivientes), reexaminados después de un promedio de 5 años, 24% tuvieron severa discapacidad, 25% tuvieron debilidad muscular residual significativa, y 84% tuvieron una calidad de vida anormal.
Medidas generales y manifestaciones extramusculares.
Además de la terapia con drogas, hay una variedad de otras importantes consideraciones en el tratamiento de las miopatías inflamatorias. Ellas incluyen la iniciación de un programa de ejercicios bajo la supervisión de un terapista físico; los pasos para prevenir la broncoaspiración en pacientes con disfunción esofágica; consejos acerca de la necesidad (para pacientes con DM) de evitar la luz ultravioleta, y profilaxis de la osteoporosis y de las infecciones oportunistas.
Ejercicio.
La terapia física y la rehabilitación deben comenzar temprano en el curso del tratamiento. Se deben realizar ejercicios de movilización activa y pasiva para prevenir contracturas articulares. Cuidado de los sitios de presión por la posibilidad de lesiones de decúbito.
Ejercicios isométricos y de resistencia deben comenzarse tan pronto el paciente haya recuperado las fuerzas y pueda realizarlos.
Riesgo de aspiración.
Los pacientes con disfagia debido a disfunción del músculo cricofaringeo están en riesgo de aspiración. Varias intervenciones pueden reducir el riesgo:
· Consulta a fonoaudiología para consejos y precaución por riesgo de aspiración.
· Elevación de la cabecera de la cama. Dietas semi-sólidas.
· Una sonda de alimentación nasogástrica puede ayudar a protejer la vía aérea en pacientes con severa disfagia.
· Los pacientes con miositis y superposición con esclerodermia pueden desarrollar esofagitis por reflujo debido a incompetencia del esfínter esofágico inferior. El tratamiento con inhibidores de la bomba de protones disminuye el riesgo de estenosis pépticas y síntomas de reflujo.
· Evitar la luz del sol. Muchos pacientes tienen fotosensibilidad cuando se los expone a la luz del sol. Para evitarlo deben usar cremas pantalla y en lo posible evitar la exposición a la luz solar.
Prevención de osteoporosis.
Todos los pacientes tratados con altas dosis de corticosteroides están en riesgo de osteoporosis inducida por corticosteroides. Así, todos los pacientes que se embarquen en tratamientos a largo plazo con corticosteroides, son candidatos a la terapia antireabsortiva para evitar la pérdida de hueso.
Infecciones oportunistas.
Debido a la alta dosis de corticosteroides y otras medicaciones inmunosupresoras existe riesgo de infección oportunista.
Pneumocystis jirovecii, un patógeno común en huéspedes inmunosuprimidos pueden causar infecciones letales.
En pacientes tratados con altas dosis de prednisona y otros agentes inmunosupresores se recomienda profilaxis contra Pneumocystis con TMS/SMZ (doble dosis) (160 mg de trimetoprima y 800 mg de sulfametoxazol) por día. Dado que la trimetoprima y el metotrexato son ambos anti-fólicos pueden producir supresión de médula ósea significativa.
Cáncer en DM y PM.
Incidencia de cáncer:
La incidencia de cáncer está aumentada 5 a 7 veces en pacientes con miopatías inflamatorias cuando se los compara con la población general.
Factores de riesgo.
La DM tiene más riesgo de cáncer que la PM. Otros factores de riesgo incluyen:
· Evidencias de daño capilar en la biopsia muscular.
· DM complicada con necrosis cutánea del tronco.
· Vasculitis cutánea leucocitoclástica.
· Edad avanzada al momento del diagnóstico de DM o PM.
· DM complicada con necrosis cutánea del tronco.
· Vasculitis cutánea leucocitoclástica.
· Edad avanzada al momento del diagnóstico de DM o PM.
En un reporte, 11 de 23 pacientes (48%) con DM o PM mayores de 65 años tuvieron cáncer, comparados con 9% de los pacientes con menos de 65 años. Diez de las once neoplasias en el grupo de edad avanzada ocurrieron en pacientes con DM y 1 en PM.
Autoanticuerpos séricos
Autoanticuerpos séricos
“Miositis asociada a cáncer” en adultos ha estado asociada con un autoanticuerpo (anti-p155/p140 o anti-p155). Este autoanticuerpo se ha visto asociado también a DM en general, a DM juevenil, pero la frecuencia en pacientes con enfermedades malignas parece ser mayor que en otros grupos.
Tipos de cáncer.
Tipos de cáncer.
Adenocarcinoma de cuello uterino, pulmón, ovario, páncreas, vejiga, y estómago dan cuenta de 70% de los cánceres asociados a miopatías inflamatorias.
Marcadores tumorales.
Marcadores tumorales.
Los marcadores tumorales pueden proveer la pista de la presencia de una neoplasia oculta en pacientes con DM o PM. Estos marcadores tumorales incluyen CA125, CA19,-9, y CEA.
Screening para cáncer.
La investigación de pacientes con miopatías inflamatorias para cáncer debe ser guiada por 3 principios:
Screening para cáncer.
La investigación de pacientes con miopatías inflamatorias para cáncer debe ser guiada por 3 principios:
· Un interrogatorio exhaustivo y un prolijo examen físico, así como análisis de laboratorio de rutina son esenciales para hacer el diagnóstico de cáncer.
· Los tests de screening que correspondan según la edad (por ejemplo mamografía, y colonoscopía) son fundamentales en pacientes con DM o PM.
· Tests adicionales como TAC tóraco-abdómino-pélvica de rutina, se recomiendan para pacientes con aumento del riesgo de cáncer.
Todos los pacientes adultos con reciente diagnóstico de DM o PM deben someterse a un completo examen físico con exámenes de mama, rectal, y ginecológico.
Evaluación de laboratorio en screening de cáncer.
· Hemograma completo.
· VSG y PCR.
· Análisis clínicos de rutina.
· CA125 y CA19-9.
· PSA.
· Investigación de sangre oculta en materia fecal.
Evaluación con imágenes
El examen por imágenes debe incluir TAC tóraco-abdómino-pélvica, y algunos expertos recomiendan eco transvaginal para mujeres con DM.
Repetición del screening
El riesgo de que aparezca un cáncer es mayor en el primer año (6 veces más riesgo) de diagnosticada la miopatía inflamatoria, disminuyendo a los 2 años (2,5 veces más riesgo).
Esto indica que debe repetirse el screening los primeros 2 a 3 años años, aunque parece razonable vigilar la aparición de cualquier síntoma que oriente hacia alguna investigación especial durante toda la vida.
Hay raros casos de cáncer de ovario que aparecen 5 años después del diagnóstico de DM.
Tratamiento de los casos recurrentes y resistentes de DM y PM.
Enfermedad recurrrente.
Esto indica que debe repetirse el screening los primeros 2 a 3 años años, aunque parece razonable vigilar la aparición de cualquier síntoma que oriente hacia alguna investigación especial durante toda la vida.
Hay raros casos de cáncer de ovario que aparecen 5 años después del diagnóstico de DM.
Tratamiento de los casos recurrentes y resistentes de DM y PM.
Enfermedad recurrrente.
Después de lograr control de la enfermedad con el tratamiento, algunos pacientes experimentan recurrencias (brotes) durante o después del período de descenso de la dosis.
Enfermedad resistente.
Enfermedad resistente.
En otros pacientes, la enfermedad no responde totalmente a la inducción de la remisión con la terapia convencional inicial, por lo que deben considerarse otras alternativas terapéuticas.
Tratamiento de la enfermedad recurrente.
Tratamiento de la enfermedad recurrente.
La suspensión total del tratamiento no tiene éxito en la mayoría de los pacientes.
Si el brote o recidiva se produce ctomando más de 10 mg de prednisona, se puede hacer 2 cosas: 1) agregado de drogas ahorradoras de corticosteroides (metotrexato o azatioprina) si ya no los estaba recibiendo, o 2) si el paciente ya estaba con azatioprina o metotrexato, recomenzar con prednisona 1 mg/kg/día para restablecer nuevamente el control de la enfermedad.
Si el brote se produce tomando 10 mg de prednisona o menos por día, se puede hacer 2 cosas que no son mutuamente excluyentes: 1) aumentar prednisona a la menor dosis requerida para restablecer el control. La dosis mínima usual es 20 mg/día. 2) Aumentar azatioprina o metotrexato a dosis mayores.
Una vez restaurado el control de la enfermedad, se sugiere descenso más lento que el descenso que ocasionó el brote. Algunos pacientes son mantenidos con dosis bajas (prednisona 5 mg/día durante 1 año o más)
Tratamiento de la enfermedad resistente.
Si el brote o recidiva se produce ctomando más de 10 mg de prednisona, se puede hacer 2 cosas: 1) agregado de drogas ahorradoras de corticosteroides (metotrexato o azatioprina) si ya no los estaba recibiendo, o 2) si el paciente ya estaba con azatioprina o metotrexato, recomenzar con prednisona 1 mg/kg/día para restablecer nuevamente el control de la enfermedad.
Si el brote se produce tomando 10 mg de prednisona o menos por día, se puede hacer 2 cosas que no son mutuamente excluyentes: 1) aumentar prednisona a la menor dosis requerida para restablecer el control. La dosis mínima usual es 20 mg/día. 2) Aumentar azatioprina o metotrexato a dosis mayores.
Una vez restaurado el control de la enfermedad, se sugiere descenso más lento que el descenso que ocasionó el brote. Algunos pacientes son mantenidos con dosis bajas (prednisona 5 mg/día durante 1 año o más)
Tratamiento de la enfermedad resistente.
Hay múltiples opciones para estos pacientes que no responden a corticosteroides, azatioprina y metotrexato.
Para ellos se han utilizado el rituximab (anti-CD20), la inmunoglobulina intravenosa, inhibidores de la calcineurina (ciclosporina y tacrolimus), ciclofosfamida, clorambucilo, inhibidores del factor de necrosis tumoral (TNF) etanercept e infliximab.
Rash refractario.
Para ellos se han utilizado el rituximab (anti-CD20), la inmunoglobulina intravenosa, inhibidores de la calcineurina (ciclosporina y tacrolimus), ciclofosfamida, clorambucilo, inhibidores del factor de necrosis tumoral (TNF) etanercept e infliximab.
Rash refractario.
Se puede usar tacrolimus tópico, antimaláricos (hidroxicloroquina, cloroquina), y micofenolato mofetil.
Fuente:
Clinical manifestations and diagnosis of adult dermatomyositis and polymyositis
Author Marc L Miller, MD
Initial treatment of dermatomyositis and polymyositis in adults
Authors Marc L Miller, MDStacy A Rudnicki, MD
Malignancy in dermatomyositis and polymyositis
Author Marc L Miller, MD
UpToDate
Bibliografía
1) Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 2003; 362:971.
2) Plotz, PH, Dalakas, M, Leff, RL, et al. Current concepts in the idiopathic inflammatory myopathies: Polymyositis, dermatomyositis and related disorders. Ann Intern Med 1989; 111:143.
3) Bohan, A, Peter, JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292:403.
4) Bohan, A, Peter, JB, Bowman, RL, Pearson, CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255.
5) Tymms, KE, Webb, J. Dermatopolymyositis and other connective tissue diseases: A review of 105 cases. J Rheumatol 1985; 12:1140.
6) Shamim, EA, Rider, LG, Pandey, JP, et al. Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican Mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis Rheum 2002; 46:1885.
7) O'Hanlon, TP, Carrick, DM, Targoff, IN, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine (Baltimore) 2006; 85:111.
8) Van Der Meulen, MF, Bronner, IM, Hoogendijk, JE, et al. Polymyositis: An overdiagnosed entity. Neurology 2003; 61:316.
9) Troyanov, Y, Targoff, IN, Tremblay, JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005; 84:231.
10) Hoogendijk, JE, Amato, AA, Lecky, BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14:337.
11) Griggs, RC, Askanas, V, DiMauro, S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995; 38:705.
12) Kissel, JT, Mendell, JR, Rammohan, KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 1986; 314:329.
13) Goebels, N, Michaelis, D, Engelhardt, M, et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest 1996; 97:2905.
14) Hohlfeld, R, Engel, AG. The immunobiology of muscle. Immunol Today 1994; 15:269.
15) Greenberg, SA, Amato, AA. Uncertainties in the pathogenesis of DM. Curr Opin Neurol 2004;13;356.
16) Greenberg, SA, Pinkus, JL, Pinkus, GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005; 57:664.
17) Emslie-Smith, AM, Arahata, K, Engel, AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol 1989; 20:224.
18) Lundberg, IE. Idiopathic inflammatory myopathies: why do the muscles become weak?. Curr Opin Rheumatol 2001; 13:457.
19) Sekiguchi, K, Kanda, F, Oishi, K, et al. HLA typing in focal myositis. J Neurol Sci 2004; 227:21.
20) Stahl, NI, Klippel, JH, Decker, JL. A cutaneous lesion associated with myositis. Ann Intern Med 1979; 91:577.
21) Yamamoto, T, Nishioka, K. Flagellate erythema. Int J Dermatol 2006; 45:627.
22) Kovacs, SO, Kovacs, SC. Dermatomyositis. J Am Acad Dermatol 1998; 39:899.
23) de Merieux, P, Verity, MA, Clements, PJ, Paulus, HE. Esophageal abnormalities and dysphagia in polymyositis and dermatomyositis. Arthritis Rheum 1983; 26:961.
24) Marie, I, Hatron, PY, Levesque, H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 1999; 78:139.
25) Denbow, CE, Lie, JT, Tancredi, RG, Bunch, TW. Cardiac involvement in polymyositis. Arthritis Rheum 1979; 22:1088.
26) Larca, LJ, Coppola , JT, Honig, S. Creatine kinase MB isoenzyme in dermatomyositis: A noncardiac source. Ann Intern Med 1981; 94:341.
27) Badsha, H, Gunes, B, Grossman, J, Brahn, E. Troponin I assessment of cardiac involvement in patients with connective tissue disease and an elevated creatine kinase MB isoform. Report of four cases and review of the literature. J Clin Rheumatol 1997; 3:131.
28) Targoff, IN. Myositis specific autoantibodies. Curr Rheumatol Rep 2006; 8:196.
29) Betteridge, Z, Gunawardena, H, North, J, et al. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 2007; 46:1005.
30) Gerami, P, Schope, JM, McDonald, L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol 2006; 54:597.
31) Euwer, RL, Sontheimer, RD. Amyopathic dermatomyositis (dermatomyositis sine myositis). Presentation of six new cases and review of the literature. J Am Acad Dermatol 1991; 24:959.
32) Stonecipher, MR, Jorizzo, JL, White, WL, et al. Cutaneous changes of dermatomyositis in patients with normal muscle enzymes: dermatomyositis sine myositis? J Am Acad Dermatol 1993; 28:951.
33) Sato, S, Hirakata, M, Kuwana, M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52:1571.
34) Ringel, RA, Brick, JE, Brick, JF, et al. Muscle involvement in the scleroderma syndromes. Arch Intern Med 1990; 150:2550.
35) Reichlin, M, Arnett, FC. Multiplicity of antibodies in myositis sera. Arthritis Rheum 1984; 27:1150.
36) Drake, LA, Dinehart, SM, Farmer, ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
37) Nimelstein, SH, Brody, S, McShane, D, Holman, HR. Mixed connective tissue disease: a subsequent evaluation of the original 25 patients. Medicine (Baltimore) 1980; 59:239.
38) Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositis – specific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
39) Plotz, PH, Rider, LG, Targoff, IN, et al. Myositis: Immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med 1995; 122:715.
40) Brouwer, R, Hengstman, GJ, Vree Egberts, W, et al. Autoantibody profiles in thesera of European patients with myositis. Ann Rheum Dis 2001; 60:116.
41) Stone, KB, Oddis, CV, Fertig, N, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007; 56:3125.
42) Seelig, HP, Moosbrugger, I, Ehlfeld, H, et al. The major dermatomyositis-specific autoantigen is a presumed helicase involved in transcriptional activation. Arthritis Rheum 1995; 38:1389.
43) Casciola-Rosen, LA, Pluta, AF, Plotz, PH, et al. The DNA mismatch repair enzyme PMS1 is a myositis-specific autoantigen. Arthritis Rheum 2001; 44:389.
44) Jablonska, S, Blaszyk, M. Scleromyositis (scleroderma/polimyositis overlap) is an entity. J Eur Acad Dermatol Venereol 2004; 18:265.
45) Targoff, IN, Mamyrova, G, Trieu, EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 2006; 54:3682.
46) Kaji, K, Fujimoto, M, Hasegawa, M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46:25.
47) Mozaffar, T, Pestronk, A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472.
48) Greenberg, SA, Sanoudou, D, Haslett, JN, et al. Molecular profiles of inflammatory myopathies. Neurology 2002; 59:1170.
49) Miller, T, Al-Lozi, MT, Lopate, G, Pestronk, A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002; 73:420.
50) Dourmishev, LA, Wollina, U. Dermatomyositis: immunopathologic study of skin lesions. Acta Dermatovenerol Alp Panonica Adriat 2006; 15:45.
51) O'Connell, MJ, Powell, T, Brennan, D, et al. Whole-body MR imaging in the diagnosis of polymyositis. AJR Am J Roentgenol 2002; 179:967.
52) Dorph, C, Nennesmo, I, Lundberg, IE. Percutaneous conchotome muscle biopsy. A useful diagnostic and assessment tool. J Rheumatol 2001; 28:1591.
53) Reimers, CD, Finkenstaedt, M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 4:475.
54) Park, JH, Olsen, NJ, King, L, et al. MRI and P-31 magnetic resonance spectroscopy detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum 1995; 38:68.
55) Plotz, PH. Not myositis. A series of chance encounters. JAMA 1992; 268:2074.
56) Amato, AA, Gronseth, GS, Jackson, CE, et al. Inclusion body myositis: Clinical and pathological boundaries. Ann Neurol 1996; 40:581.
57) Dion, E, Cherin, P, Payan, C, et al. Magnetic resonance imaging criteria for distinguishing between inclusion body myositis and polymyositis. J Rheumatol 2002; 29:1897.
58) Tonin, P, Lewis, P, Servidei, S, DiMauro, S. Metabolic causes of myoglobinuria. Ann Neurol 1990; 27:181.
59) Kim, HW, Choi, JR, Jang, SJ, et al. Recurrent rhabdomyolysis and myoglobinuric acute renal failure in a patient with polymyositis. Nephrol Dial Transplant 2005; 20:2255.
60) Caccamo, DV, Keene, CY, Durhan, J, Peven, D. Fulminant rhabdomyolysis in a patient with dermatomyositis. Neurology 1993; 43:844.
61) Rider, LG, Gurley, RC, Pandey, JP. Clinical, serologic, and immunogenetic features of familial idiopathic inflammatory myopathy. Arthritis Rheum 1998; 41:710.
62) Parker, P, Chao, NJ, Ben-Ezra, J, et al. Polymyositis as a manifestation of chronic graft-versus-host disease. Medicine (Baltimore) 1996; 75:279.
63) Stevens, AM, Sullivan, KM, Nelson, JL. Polymyositis as a manifestation of chronic graft-versus-host disease. Rheumatology (Oxford) 2003; 42:34.
64) Mandl, LA, Folkerth, RD, Pick, MA, et al. Amyloid myopathy masquerading as polymyositis. J Rheumatol 2000; 27:949.
65) Bunch, TW, Worthington, JW, Combs, JJ, et al. Azathioprine with prednisone for polymyositis. Ann Intern Med 1980; 92:365.
66) Dalakas, MC, Illa, I, Dambrosia, JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993; 329:1993.
67) Miller, FW, Leitman, SF, Cronin, ME, et al. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med 1992; 326:1380.
68) Troyanov, Y, Targoff, IN, Tremblay, JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005; 84:231.
69) Hoogendijk, JE, Amato, AA, Lecky, BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14:337.
70) Isenberg, DA, Allen, E, Farewell, V, et al. International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford) 2004; 43:49.
71) Joffe, MM, Love, LA, Leff, RL, et al. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379.
72) Fafalak, RG, Peterson, MGE, Kagen, LJ. Strength in polymyositis and dermatomyositis: Best outcome in patients treated early. J Rheumatol 1994; 21:643.
73) Carpenter, JR, Bunch, TW, Engel, AG, O'Brien, PC. Survival in polymyositis: Corticosteroids and risk factors. J Rheumatol 1977; 4:207.
74) Danko, K, Ponyi, A, Constantin, T, et al. Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 2004; 83:35.
75) Maugars, YM, Berthelot, JM, Abbas, AA, et al. Long-term prognosis of 69 patients with dermatomyositis or polymyositis. Clin Exp Rheumatol 1996; 14:263.
76) Marie, I, Hatron, PY, Levesque, H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 1999; 78:139.
77) Lundberg, IE, Forbess, CJ. Mortality in idiopathic inflammatory myopathies. Clin Exp Rheum 2008; 26 (Suppl 51):S109.
78) Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositis-specific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
79) Plotz, PH, Rider, LG, Targoff IN, et al. Myositis: immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med 1995; 122:715.
80) Stone, KB, Oddis, CV, Fertig, N, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007; 56:3125.
81) Miller, T, Al-Lozi, MT, Lopate, G, Pestronk, A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002; 73:420.
82) Kao, AH, Lacomis, D, Lucas, M, et al. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum 2004; 50:209.
83) Targoff, IN. Immune manifestations of inflammatory muscle disease. Rheum Dis Clin North Am 1994; 20:857.
84) Winkelmann, RK, Mulder, DW, Lambert, EH, et al. Course of dermatomyositis-polymyositis: Comparison of untreated and cortisone-treated patients. Mayo Clin Proc 1968; 43:545.
85) Drake, LA, Dinehart, SM, Farmer, ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
86) Hoffman, GS, Franck, WA, Raddatz, DA, Stallones, L. Presentation, treatment, and prognosis of idiopathic inflammatory muscle disease in a rural hospital. Am J Med 1983; 75:433.
87) Robinson, LR. AAEM case report #22: polymyositis. Muscle Nerve 1991; 14:310.
88) Sigurgeirsson, B, Lindelof, B, Edhag, O, Allander, E. Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med 1992; 326:363.
89) Bunch, TW. Prednisone and azathioprine for polymyositis. Long-term followup. Arthritis Rheum 1981; 24:45.
90) Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 2003; 362:971.
91) Amato, AA, Griggs, RC. Treatment of idiopathic inflammatory myopathies. Curr Opin Neurol 2003; 16:569.
92) Kissel, JT, Levy, RJ, Mendell, JR, Griggs, RC. Azathioprine toxicity in neuromuscular disease. Neurology 1986; 36:35.
93) Giannini, M, Callen, JP. Treatment of dermatomyositis with methotrexate and prednisone. Arch Dermatol 1979; 115:1251.
94) Newman, ED, Scott, DW. The use of low-dose oral methotrexate in the treatment of polymyositis and dermatomyositis. J Clin Rheumatol 1995; 1:99.
95) Metzger, AL, Bohan, A, Goldberg, LS, et al. Polymyositis and dermatomyositis: Combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182.
96) Bronner, IM, van der, Meulen MF, de Visser, M, et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis 2006; 65:1456.
97) Alexanderson, H, Lundberg, IE. The role of exercise in the rehabilitation of idiopathic inflammatory myopathies. Curr Opin Rheumatol 2005; 17:164.
98) Hicks, JE, Miller, F, Plotz, P, et al. Isometric exercise increases strength and does not produce sustained creatinine phosphokinase increases in a patient with polymyositis. J Rheumatol 1993; 20:1399.
99) Silva, CA, Sultan, SM, Isenberg, DA. Pregnancy outcome in adult-onset idiopathic inflammatory myopathy. Rheumatology (Oxford) 2003; 42:1168
100) Casciola-Rosen, L, Nagaraju, K, Plotz, P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med 2005; 201:591.
101) Levine, SM. Cancer and myositis: new insights into an old association. Curr Opin Rheumatol 2006; 18:620.
102) Furneaux, HM, Rosenblum, MK, Dalmau, J, et al. Selective expression of Purkinje-cell antigens in tumor tissue from patients with paraneoplastic cerebellar degeneration. N Engl J Med 1990; 322:1844.
103) Albert, ML, Darnell, RB. Paraneoplastic neurological degenerations: keys to tumour immunity. Nat Rev Cancer 2004; 4:36.
104) Stertz, O. Polymyositis. Berl Klin Wochenschr 1916; 53:489.
105) Barnes, BE. Dermatomyositis and malignancy. Ann Intern Med 1976; 84:68.
106) Sigurgeirsson, B, Lindelof, B, Edhag, O, Allander, E. Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med 1992; 326:363.
107) Buchbinder, R, Forbes, A, Hall, S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population-based cohort study. Ann Intern Med 2001; 134:1087.
108) Stockton, D, Doherty, VR, Brewster, DH. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer 2001; 85:41.
109) Lakhanpal, S, Bunch, TW, Ilstrup, DM, Melton, III LJ. Polymyositis-dermatomyositis and malignant lesions: Does an association exist? Mayo Clin Proc 1986; 61:645.
110) Manchul, LA, Jin, A, Pritchard, KI, et al. The frequency of malignant neoplasms in patients with polymyositis-dermatomyositis. Arch Intern Med 1985; 145:1835.
111) Tymms, KE, Webb, J. Dermatopolymyositis and other connective tissue diseases: A review of 105 cases. J Rheumatol 1985; 12:1140.
112) Callen, JP. The value of malignancy evaluation in patients with dermatomyositis. J Am Acad Dermatol 1982; 6:253.
113) Cox, NH, Lawrence, CM, Langtry, JA, Ive, FA. Dermatomyositis: disease associations and an evaluation of screening investigations for malignancy. Arch Dermatol 1990; 126:61.
114) Zantos, D, Zhang, Y, Felson, D. The overall and temporal association of cancer with polymyositis and dermatomyositis. J Rheumatol 1994; 21:1855.
115) Urbano-Marquez, A, Casademont, J, Grau, JM. Polymyositis/dermatomyositis: The current position. Ann Rheum Dis 1991; 50:191.
116) Basset-Seguin, N, Roujeau, J, Gherardi, R, et al. Prognostic factors and predictive signs of malignancy in adult dermatomyositis. Arch Dermatol 1990; 126:633.
117) Hunger, RE, Durr, C, Brand, CU. Cutaneous leukocytoclastic vasculitis in dermatomyositis suggests malignancy. Dermatology 2001; 202:123.
118) Marie, I, Hatron, PY, Levesque, H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 1999; 78:139.
119) Chinoy, H, Fertig, N, Oddis, CV, et al. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007; 66:1345.
120) Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 2006;54:3682.
121) Kaji, K, Fujimoto, M, Hasegawa, M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46:25.
122) Hill, CL, Zhang, Y, Sigurgeirsson, B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 2001; 357:96.
123) Bivalacqua, TJ, Alphs, H, Aksentijevich, I, et al. Paraneoplastic polyarthritis from non-small-cell lung cancer metastatic to the bladder. J Clin Oncol 2007; 25:2621.
124) Ang, P, Sugeng, MW, Chua, SH. Classical and amyopathic dermatomyositis seen at the National Skin Centre of Singapore: a 3-year retrospective review of their clinical characteristics and association with malignancy. Ann Acad Med Singapore 2000; 29:219.
125) Amoura, Z, Duhaut, P, Huong, DL, et al. Tumor antigen markers for the detection of solid cancers in inflammatory myopathies. Cancer Epidemiol Biomarkers Prev 2005; 14:1279.
126) Drake, LA, Dinehart, SM, Farmer, ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
127) Sparsa, A, Liozon, E, Herrmann, F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol 2002; 138:885.
128) Provost, TT, Flynn, JA. Dermatomyositis. In: Cutaneous Medicine, Provost, TT, Flynn, JA (Eds). Hamilton, Ontario, B.C. Decker Inc, 2001, p. 82-103.
129) Chow, WH, Gridley, G, Mellemkjaer, L, et al. Cancer risk following polymyositis and dermatomyositis: A nationwide cohort study in Denmark. Cancer Causes Control 1995; 6:9.
Fuente:
Clinical manifestations and diagnosis of adult dermatomyositis and polymyositis
Author Marc L Miller, MD
Initial treatment of dermatomyositis and polymyositis in adults
Authors Marc L Miller, MDStacy A Rudnicki, MD
Malignancy in dermatomyositis and polymyositis
Author Marc L Miller, MD
UpToDate
Bibliografía
1) Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 2003; 362:971.
2) Plotz, PH, Dalakas, M, Leff, RL, et al. Current concepts in the idiopathic inflammatory myopathies: Polymyositis, dermatomyositis and related disorders. Ann Intern Med 1989; 111:143.
3) Bohan, A, Peter, JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292:403.
4) Bohan, A, Peter, JB, Bowman, RL, Pearson, CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255.
5) Tymms, KE, Webb, J. Dermatopolymyositis and other connective tissue diseases: A review of 105 cases. J Rheumatol 1985; 12:1140.
6) Shamim, EA, Rider, LG, Pandey, JP, et al. Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican Mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis Rheum 2002; 46:1885.
7) O'Hanlon, TP, Carrick, DM, Targoff, IN, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine (Baltimore) 2006; 85:111.
8) Van Der Meulen, MF, Bronner, IM, Hoogendijk, JE, et al. Polymyositis: An overdiagnosed entity. Neurology 2003; 61:316.
9) Troyanov, Y, Targoff, IN, Tremblay, JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005; 84:231.
10) Hoogendijk, JE, Amato, AA, Lecky, BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14:337.
11) Griggs, RC, Askanas, V, DiMauro, S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995; 38:705.
12) Kissel, JT, Mendell, JR, Rammohan, KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 1986; 314:329.
13) Goebels, N, Michaelis, D, Engelhardt, M, et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest 1996; 97:2905.
14) Hohlfeld, R, Engel, AG. The immunobiology of muscle. Immunol Today 1994; 15:269.
15) Greenberg, SA, Amato, AA. Uncertainties in the pathogenesis of DM. Curr Opin Neurol 2004;13;356.
16) Greenberg, SA, Pinkus, JL, Pinkus, GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005; 57:664.
17) Emslie-Smith, AM, Arahata, K, Engel, AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol 1989; 20:224.
18) Lundberg, IE. Idiopathic inflammatory myopathies: why do the muscles become weak?. Curr Opin Rheumatol 2001; 13:457.
19) Sekiguchi, K, Kanda, F, Oishi, K, et al. HLA typing in focal myositis. J Neurol Sci 2004; 227:21.
20) Stahl, NI, Klippel, JH, Decker, JL. A cutaneous lesion associated with myositis. Ann Intern Med 1979; 91:577.
21) Yamamoto, T, Nishioka, K. Flagellate erythema. Int J Dermatol 2006; 45:627.
22) Kovacs, SO, Kovacs, SC. Dermatomyositis. J Am Acad Dermatol 1998; 39:899.
23) de Merieux, P, Verity, MA, Clements, PJ, Paulus, HE. Esophageal abnormalities and dysphagia in polymyositis and dermatomyositis. Arthritis Rheum 1983; 26:961.
24) Marie, I, Hatron, PY, Levesque, H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 1999; 78:139.
25) Denbow, CE, Lie, JT, Tancredi, RG, Bunch, TW. Cardiac involvement in polymyositis. Arthritis Rheum 1979; 22:1088.
26) Larca, LJ, Coppola , JT, Honig, S. Creatine kinase MB isoenzyme in dermatomyositis: A noncardiac source. Ann Intern Med 1981; 94:341.
27) Badsha, H, Gunes, B, Grossman, J, Brahn, E. Troponin I assessment of cardiac involvement in patients with connective tissue disease and an elevated creatine kinase MB isoform. Report of four cases and review of the literature. J Clin Rheumatol 1997; 3:131.
28) Targoff, IN. Myositis specific autoantibodies. Curr Rheumatol Rep 2006; 8:196.
29) Betteridge, Z, Gunawardena, H, North, J, et al. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 2007; 46:1005.
30) Gerami, P, Schope, JM, McDonald, L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol 2006; 54:597.
31) Euwer, RL, Sontheimer, RD. Amyopathic dermatomyositis (dermatomyositis sine myositis). Presentation of six new cases and review of the literature. J Am Acad Dermatol 1991; 24:959.
32) Stonecipher, MR, Jorizzo, JL, White, WL, et al. Cutaneous changes of dermatomyositis in patients with normal muscle enzymes: dermatomyositis sine myositis? J Am Acad Dermatol 1993; 28:951.
33) Sato, S, Hirakata, M, Kuwana, M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52:1571.
34) Ringel, RA, Brick, JE, Brick, JF, et al. Muscle involvement in the scleroderma syndromes. Arch Intern Med 1990; 150:2550.
35) Reichlin, M, Arnett, FC. Multiplicity of antibodies in myositis sera. Arthritis Rheum 1984; 27:1150.
36) Drake, LA, Dinehart, SM, Farmer, ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
37) Nimelstein, SH, Brody, S, McShane, D, Holman, HR. Mixed connective tissue disease: a subsequent evaluation of the original 25 patients. Medicine (Baltimore) 1980; 59:239.
38) Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositis – specific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
39) Plotz, PH, Rider, LG, Targoff, IN, et al. Myositis: Immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med 1995; 122:715.
40) Brouwer, R, Hengstman, GJ, Vree Egberts, W, et al. Autoantibody profiles in thesera of European patients with myositis. Ann Rheum Dis 2001; 60:116.
41) Stone, KB, Oddis, CV, Fertig, N, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007; 56:3125.
42) Seelig, HP, Moosbrugger, I, Ehlfeld, H, et al. The major dermatomyositis-specific autoantigen is a presumed helicase involved in transcriptional activation. Arthritis Rheum 1995; 38:1389.
43) Casciola-Rosen, LA, Pluta, AF, Plotz, PH, et al. The DNA mismatch repair enzyme PMS1 is a myositis-specific autoantigen. Arthritis Rheum 2001; 44:389.
44) Jablonska, S, Blaszyk, M. Scleromyositis (scleroderma/polimyositis overlap) is an entity. J Eur Acad Dermatol Venereol 2004; 18:265.
45) Targoff, IN, Mamyrova, G, Trieu, EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 2006; 54:3682.
46) Kaji, K, Fujimoto, M, Hasegawa, M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46:25.
47) Mozaffar, T, Pestronk, A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472.
48) Greenberg, SA, Sanoudou, D, Haslett, JN, et al. Molecular profiles of inflammatory myopathies. Neurology 2002; 59:1170.
49) Miller, T, Al-Lozi, MT, Lopate, G, Pestronk, A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002; 73:420.
50) Dourmishev, LA, Wollina, U. Dermatomyositis: immunopathologic study of skin lesions. Acta Dermatovenerol Alp Panonica Adriat 2006; 15:45.
51) O'Connell, MJ, Powell, T, Brennan, D, et al. Whole-body MR imaging in the diagnosis of polymyositis. AJR Am J Roentgenol 2002; 179:967.
52) Dorph, C, Nennesmo, I, Lundberg, IE. Percutaneous conchotome muscle biopsy. A useful diagnostic and assessment tool. J Rheumatol 2001; 28:1591.
53) Reimers, CD, Finkenstaedt, M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 4:475.
54) Park, JH, Olsen, NJ, King, L, et al. MRI and P-31 magnetic resonance spectroscopy detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum 1995; 38:68.
55) Plotz, PH. Not myositis. A series of chance encounters. JAMA 1992; 268:2074.
56) Amato, AA, Gronseth, GS, Jackson, CE, et al. Inclusion body myositis: Clinical and pathological boundaries. Ann Neurol 1996; 40:581.
57) Dion, E, Cherin, P, Payan, C, et al. Magnetic resonance imaging criteria for distinguishing between inclusion body myositis and polymyositis. J Rheumatol 2002; 29:1897.
58) Tonin, P, Lewis, P, Servidei, S, DiMauro, S. Metabolic causes of myoglobinuria. Ann Neurol 1990; 27:181.
59) Kim, HW, Choi, JR, Jang, SJ, et al. Recurrent rhabdomyolysis and myoglobinuric acute renal failure in a patient with polymyositis. Nephrol Dial Transplant 2005; 20:2255.
60) Caccamo, DV, Keene, CY, Durhan, J, Peven, D. Fulminant rhabdomyolysis in a patient with dermatomyositis. Neurology 1993; 43:844.
61) Rider, LG, Gurley, RC, Pandey, JP. Clinical, serologic, and immunogenetic features of familial idiopathic inflammatory myopathy. Arthritis Rheum 1998; 41:710.
62) Parker, P, Chao, NJ, Ben-Ezra, J, et al. Polymyositis as a manifestation of chronic graft-versus-host disease. Medicine (Baltimore) 1996; 75:279.
63) Stevens, AM, Sullivan, KM, Nelson, JL. Polymyositis as a manifestation of chronic graft-versus-host disease. Rheumatology (Oxford) 2003; 42:34.
64) Mandl, LA, Folkerth, RD, Pick, MA, et al. Amyloid myopathy masquerading as polymyositis. J Rheumatol 2000; 27:949.
65) Bunch, TW, Worthington, JW, Combs, JJ, et al. Azathioprine with prednisone for polymyositis. Ann Intern Med 1980; 92:365.
66) Dalakas, MC, Illa, I, Dambrosia, JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993; 329:1993.
67) Miller, FW, Leitman, SF, Cronin, ME, et al. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med 1992; 326:1380.
68) Troyanov, Y, Targoff, IN, Tremblay, JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005; 84:231.
69) Hoogendijk, JE, Amato, AA, Lecky, BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14:337.
70) Isenberg, DA, Allen, E, Farewell, V, et al. International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford) 2004; 43:49.
71) Joffe, MM, Love, LA, Leff, RL, et al. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379.
72) Fafalak, RG, Peterson, MGE, Kagen, LJ. Strength in polymyositis and dermatomyositis: Best outcome in patients treated early. J Rheumatol 1994; 21:643.
73) Carpenter, JR, Bunch, TW, Engel, AG, O'Brien, PC. Survival in polymyositis: Corticosteroids and risk factors. J Rheumatol 1977; 4:207.
74) Danko, K, Ponyi, A, Constantin, T, et al. Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 2004; 83:35.
75) Maugars, YM, Berthelot, JM, Abbas, AA, et al. Long-term prognosis of 69 patients with dermatomyositis or polymyositis. Clin Exp Rheumatol 1996; 14:263.
76) Marie, I, Hatron, PY, Levesque, H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 1999; 78:139.
77) Lundberg, IE, Forbess, CJ. Mortality in idiopathic inflammatory myopathies. Clin Exp Rheum 2008; 26 (Suppl 51):S109.
78) Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositis-specific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
79) Plotz, PH, Rider, LG, Targoff IN, et al. Myositis: immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med 1995; 122:715.
80) Stone, KB, Oddis, CV, Fertig, N, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007; 56:3125.
81) Miller, T, Al-Lozi, MT, Lopate, G, Pestronk, A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002; 73:420.
82) Kao, AH, Lacomis, D, Lucas, M, et al. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum 2004; 50:209.
83) Targoff, IN. Immune manifestations of inflammatory muscle disease. Rheum Dis Clin North Am 1994; 20:857.
84) Winkelmann, RK, Mulder, DW, Lambert, EH, et al. Course of dermatomyositis-polymyositis: Comparison of untreated and cortisone-treated patients. Mayo Clin Proc 1968; 43:545.
85) Drake, LA, Dinehart, SM, Farmer, ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
86) Hoffman, GS, Franck, WA, Raddatz, DA, Stallones, L. Presentation, treatment, and prognosis of idiopathic inflammatory muscle disease in a rural hospital. Am J Med 1983; 75:433.
87) Robinson, LR. AAEM case report #22: polymyositis. Muscle Nerve 1991; 14:310.
88) Sigurgeirsson, B, Lindelof, B, Edhag, O, Allander, E. Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med 1992; 326:363.
89) Bunch, TW. Prednisone and azathioprine for polymyositis. Long-term followup. Arthritis Rheum 1981; 24:45.
90) Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 2003; 362:971.
91) Amato, AA, Griggs, RC. Treatment of idiopathic inflammatory myopathies. Curr Opin Neurol 2003; 16:569.
92) Kissel, JT, Levy, RJ, Mendell, JR, Griggs, RC. Azathioprine toxicity in neuromuscular disease. Neurology 1986; 36:35.
93) Giannini, M, Callen, JP. Treatment of dermatomyositis with methotrexate and prednisone. Arch Dermatol 1979; 115:1251.
94) Newman, ED, Scott, DW. The use of low-dose oral methotrexate in the treatment of polymyositis and dermatomyositis. J Clin Rheumatol 1995; 1:99.
95) Metzger, AL, Bohan, A, Goldberg, LS, et al. Polymyositis and dermatomyositis: Combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182.
96) Bronner, IM, van der, Meulen MF, de Visser, M, et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis 2006; 65:1456.
97) Alexanderson, H, Lundberg, IE. The role of exercise in the rehabilitation of idiopathic inflammatory myopathies. Curr Opin Rheumatol 2005; 17:164.
98) Hicks, JE, Miller, F, Plotz, P, et al. Isometric exercise increases strength and does not produce sustained creatinine phosphokinase increases in a patient with polymyositis. J Rheumatol 1993; 20:1399.
99) Silva, CA, Sultan, SM, Isenberg, DA. Pregnancy outcome in adult-onset idiopathic inflammatory myopathy. Rheumatology (Oxford) 2003; 42:1168
100) Casciola-Rosen, L, Nagaraju, K, Plotz, P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med 2005; 201:591.
101) Levine, SM. Cancer and myositis: new insights into an old association. Curr Opin Rheumatol 2006; 18:620.
102) Furneaux, HM, Rosenblum, MK, Dalmau, J, et al. Selective expression of Purkinje-cell antigens in tumor tissue from patients with paraneoplastic cerebellar degeneration. N Engl J Med 1990; 322:1844.
103) Albert, ML, Darnell, RB. Paraneoplastic neurological degenerations: keys to tumour immunity. Nat Rev Cancer 2004; 4:36.
104) Stertz, O. Polymyositis. Berl Klin Wochenschr 1916; 53:489.
105) Barnes, BE. Dermatomyositis and malignancy. Ann Intern Med 1976; 84:68.
106) Sigurgeirsson, B, Lindelof, B, Edhag, O, Allander, E. Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med 1992; 326:363.
107) Buchbinder, R, Forbes, A, Hall, S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population-based cohort study. Ann Intern Med 2001; 134:1087.
108) Stockton, D, Doherty, VR, Brewster, DH. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer 2001; 85:41.
109) Lakhanpal, S, Bunch, TW, Ilstrup, DM, Melton, III LJ. Polymyositis-dermatomyositis and malignant lesions: Does an association exist? Mayo Clin Proc 1986; 61:645.
110) Manchul, LA, Jin, A, Pritchard, KI, et al. The frequency of malignant neoplasms in patients with polymyositis-dermatomyositis. Arch Intern Med 1985; 145:1835.
111) Tymms, KE, Webb, J. Dermatopolymyositis and other connective tissue diseases: A review of 105 cases. J Rheumatol 1985; 12:1140.
112) Callen, JP. The value of malignancy evaluation in patients with dermatomyositis. J Am Acad Dermatol 1982; 6:253.
113) Cox, NH, Lawrence, CM, Langtry, JA, Ive, FA. Dermatomyositis: disease associations and an evaluation of screening investigations for malignancy. Arch Dermatol 1990; 126:61.
114) Zantos, D, Zhang, Y, Felson, D. The overall and temporal association of cancer with polymyositis and dermatomyositis. J Rheumatol 1994; 21:1855.
115) Urbano-Marquez, A, Casademont, J, Grau, JM. Polymyositis/dermatomyositis: The current position. Ann Rheum Dis 1991; 50:191.
116) Basset-Seguin, N, Roujeau, J, Gherardi, R, et al. Prognostic factors and predictive signs of malignancy in adult dermatomyositis. Arch Dermatol 1990; 126:633.
117) Hunger, RE, Durr, C, Brand, CU. Cutaneous leukocytoclastic vasculitis in dermatomyositis suggests malignancy. Dermatology 2001; 202:123.
118) Marie, I, Hatron, PY, Levesque, H, et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore) 1999; 78:139.
119) Chinoy, H, Fertig, N, Oddis, CV, et al. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007; 66:1345.
120) Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 2006;54:3682.
121) Kaji, K, Fujimoto, M, Hasegawa, M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46:25.
122) Hill, CL, Zhang, Y, Sigurgeirsson, B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 2001; 357:96.
123) Bivalacqua, TJ, Alphs, H, Aksentijevich, I, et al. Paraneoplastic polyarthritis from non-small-cell lung cancer metastatic to the bladder. J Clin Oncol 2007; 25:2621.
124) Ang, P, Sugeng, MW, Chua, SH. Classical and amyopathic dermatomyositis seen at the National Skin Centre of Singapore: a 3-year retrospective review of their clinical characteristics and association with malignancy. Ann Acad Med Singapore 2000; 29:219.
125) Amoura, Z, Duhaut, P, Huong, DL, et al. Tumor antigen markers for the detection of solid cancers in inflammatory myopathies. Cancer Epidemiol Biomarkers Prev 2005; 14:1279.
126) Drake, LA, Dinehart, SM, Farmer, ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
127) Sparsa, A, Liozon, E, Herrmann, F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol 2002; 138:885.
128) Provost, TT, Flynn, JA. Dermatomyositis. In: Cutaneous Medicine, Provost, TT, Flynn, JA (Eds). Hamilton, Ontario, B.C. Decker Inc, 2001, p. 82-103.
129) Chow, WH, Gridley, G, Mellemkjaer, L, et al. Cancer risk following polymyositis and dermatomyositis: A nationwide cohort study in Denmark. Cancer Causes Control 1995; 6:9.