Paciente de 16 años, sexo masculino que ingresa por
paresia de miembros inferiores y superiores. Fuerza muscular ++/+++++ en 4 extremidades
Este paciente ya tiene diagnostico confirmado de
leucosistrofia metacromatica
Refieren los familiares que el caminaba con ayuda, hace una semana
empezó con cuadros febriles, fue manejado ambulatoriamente con dx de neumonia.
Ingreso por disminución marcada de la fuerza muscular en las 4 extremidades
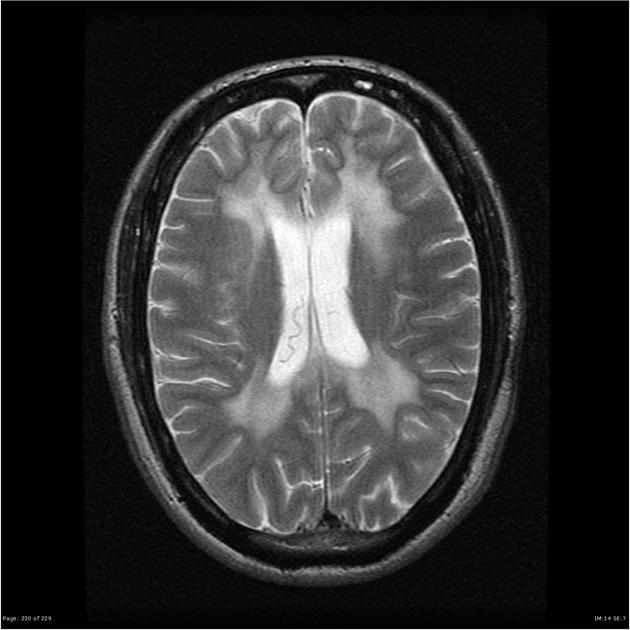
Se adjunta estudio de TC realizado al ingreso a UTI
donde se puede apreciar el compomiso
bilateral simétrico de la sustancia blanca principalmente a nivel
frontal y periventricular.

Gentileza:
Dr. Juan Pablo
Universidad Simón Bolívar.
Barranquilla. Colombia
LEUCODISTROFIA METACROMÁTICA
INTRODUCCIÓN
-
La leucodistrofia metacromática (lipidosis
sulfatide) (LDM) es una rara enfermedad de almacenamiento lisosomal autosómica recesiva que causa desmielinización progresiva del sistema
nervioso central y periférico.
ETIOLOGÍA
La leucodistrofia metacromática (LDM) es causada por
la actividad deficiente de la arilsulfatasa A, como resultado de, en casi todos
los casos, las mutaciones en el gen de la arilsulfatasa (gen ARSA). En algunos
pacientes, la LDM es causada por una deficiencia proteína activadora de
esfingolípido SAP-B (saposina B), que normalmente estimula la degradación de
sulfátidos por ARSA [ 1 ]. Esta forma variante de LDM está causada por
mutaciones en el gen de prosaposina (gen PSAP).
La arilsulfatasa (ARSA) es responsable de la desulfatación
de sulfato de cerebrosido, uno de los principales glicolípidos de la mielina.
Como resultado, la disminución de la actividad ARSA conduce a la acumulación de
sulfato de cerebrosido en el sistema central nervioso, los nervios periféricos,
los riñones y otros órganos viscerales. La acumulación de sulfato de
cerebrosido destruye la oligodendroglia y las células de Schwann, causando
desmielinización central y periférica. La microscopía electrónica muestra
líneas engrosadas en los verticilos de mielina e inclusiones lamelares de
sulfátidos en las células de Schwann [ 2 ].
Genética
- Al menos 60 mutaciones en el
gen ARSA se han descrito en LDM. Dos alelos, A y I, juntos representan
aproximadamente el 50 por ciento de los casos [ 3,4 ]. Estos alelos contribuyen
a las diferentes expresiones clínicas de la enfermedad [ 3 ].
● homocigosis para el alelo I se asocia con la
actividad ARSA residual muy baja o indetectable y la aparición infantil tardía;
heterocigotos compuestos (con el otro alelo desconocido) también parecen tener
un inicio infantil tardía.
● homocigosis para el alelo A se asocia con la
actividad ARSA residual baja, pero detectable y los juveniles o adultos formas
de inicio; heterocigotos compuestos tienen un comienzo tardío de la enfermedad.
● La presencia de ambos alelos se asocia con inicio
juvenil.
EPIDEMIOLOGÍA
La prevalencia de la leucodistrofia metacromática varía de 1: 40.000 a 1: 100.000 en las poblaciones
europeas y norteamericanas del Norte [ 8,9 ]. Sin embargo, una mayor
prevalencia se ha encontrado en ciertos grupos, incluyendo Judíos Habbanite en
Israel, los árabes que viven en Israel, y los indios Navajo en los Estados
Unidos [ 10-12 ].
MANIFESTACIONES CLÍNICAS
Tres subtipos principales de leucodistrofia
metacromática se distinguen
principalmente por la edad de inicio de la enfermedad [ 3 ]:
● inicio infantil tardío (de 6 meses a 2 años)
● inicio juvenil (edad 3 a 16 años)
● aparición en el adulto (edad mayor a 16 años)
La neuropatía periférica se produce en todas las
formas y puede ser una característica de presentación, en particular en la
forma infantil tardía ( tabla 1 ) [ 14-16 ]. La participación de la vesícula
biliar es común con manifestaciones que incluyen pólipos hiperplásicos y un
aumento del riesgo probable de carcinoma de la vesícula biliar [ 17-21 ].
INICIO INFANTIL TARDÍO - La
forma infantil tardía de LDM aparece típicamente en seis meses a dos años de
edad, aunque el inicio hasta cuatro años de edad se considera infantil tardía
por algunos investigadores [ 22 ]. Los primeros signos incluyen la regresión de
las habilidades motoras, dificultades de la marcha, convulsiones, ataxia,
hipotonía, respuestas plantares extensoras, y atrofia óptica [ 22,23 ]. Los
reflejos tendinosos profundos son a veces reducidos o ausentes, lo que refleja
una neuropatía periférica. El pronóstico es peor que las formas de aparición
tardía de LDM; la progresión a la muerte se produce normalmente dentro de cinco
a seis años.
FORMAS JUVENIL
- La forma de aparición juvenil
de LDM es heterogénea en la presentación. Algunos niños presentan entre cuatro
y seis años de edad (juvenil temprana) con, deterioro intelectual, problemas de
conducta, trastornos de la marcha, ataxia, signos de motoneurona superior, y
una neuropatía periférica [ 22,24 ]. Se pueden producir convulsiones, y la
progresión a la muerte ocurre dentro de los seis años de su inicio. Otro grupo
de niños presentes entre 6 y 16 años de edad ( juvenil tardía) con cambios de
comportamiento y deterioro intelectual y, en muchos casos, convulsiones. La progresión es más lenta, y
estos niños pueden sobrevivir hasta la edad adulta temprana.
APARICIÓN EN EL ADULTO DE LDM (17 años o más) es por
lo general anunciada por demencia y dificultades de comportamiento, y una
minoría sustancial se presente con
neuropatía, psicosis, esquizofrenia, o convulsiones [ 22 ]. Atrofia
óptica También se ha informado [ 25 ].
En las tres formas de presentación se pueden observar los
siguientes síntomas y signos comunes
- Movimiento muscular anormal
- Problemas de comportamiento
- Disminución de la función mental
- Disminución del tono muscular
- Dificultad para caminar
- Dificultad para comer o alimentarse
- Caídas frecuentes
- Incontinencia
- Irritabilidad
- Pérdida de control muscular
- Convulsiones
- Dificultad para hablar
- Dificultad para tragar



INVESTIGACIONES
- En la aparición tardía de LDM,
estudios de conducción nerviosa muestran una marcada desaceleración. Los
potenciales sensitivos se ven afectados más temprano y con mayor severidad que
son respuestas motoras [ 26,27 ]. Hay desmielinización segmentaria con
material metacromático en las células de
Schwann y los macrófagos en la biopsia del nervio periférico [ 28,29 ].
La RMN cerebral revela lesiones de sustancia blanca
simétricas con predominio periventricular en forma temprana de la enfermedad y atrofia cortical en las formas posteriores (
algoritmo 1 ) [ 30 ]. Un estudio retrospectivo informó que los niveles de
N-acetil aspartato (NAA) medidos por espectroscopia de resonancia magnética
pueden ser útiles en ensayos terapéuticos como un biomarcador de progresión de
LDM [ 31 ], pero no se ha establecido la
utilidad de los niveles de NAA en la práctica clínica.
DIAGNÓSTICO
- El cuadro clínico combinado con
una reducción de velocidad de conducción nerviosa y las concentraciones de
proteínas de líquido cefalorraquídeo elevadas debe sugerir el diagnóstico de la
leucodistrofia metacromática (LDM).
El diagnóstico de LDM se establece mediante la
demostración de la deficiencia de arilsulfatasa, un gen deficiente (gen ARSA) en los leucocitos o fibroblastos de piel
cultivados. Los valores varían típicamente de indetectable a menos del 10 por
ciento de los valores normales. Sin embargo, el diagnóstico basado únicamente
en la actividad de ARSA se complica por la existencia de pseudodeficiencia de ARSA (presente en aproximadamente el uno por
ciento de la población general). Como resultado, la detección de la presencia
de los alelos pseudodeficiencia es importante cuando se detectan niveles bajos
pero no ausentes en las pruebas prenatales o proyección de familiares
asintomáticos [ 7 ].
TRATAMIENTO
- No hay tratamiento curativo
está actualmente disponible para leucodistrofia metacromática. El trasplante
alogénico de células hematopoyéticas ha
ralentizado la progresión de la enfermedad en algunos pacientes [ 32-35 ].
La evidencia preliminar sugiere que la terapia
génica, trasplante de células madre hematopoyéticas en combinación con la
terapia génica, y la terapia de reemplazo de enzimas son las opciones de
tratamiento prometedoras [ 36 ].
RESUMEN
● leucodistrofia metacromática (LDM) es una
enfermedad de almacenamiento lisosomal autosómica recesiva causada por
mutaciones en el gen de la arilsulfatasa (gen ARSA). En algunos pacientes, una
forma variante de LDM está causada por mutaciones en el gen de prosaposina (gen
PSAP).
● La prevalencia de LDM varía de 1: 40.000 a 1:
100.000 en las poblaciones del norte de Europa y Norteamérica.
● Tres subtipos principales de LDM se distinguen
principalmente por la edad al inicio de la enfermedad, e incluyen formas
finales infantil, juvenil y adulto.
• La forma infantil tardía de LDM presenta desde la
edad de seis meses a dos años; los primeros signos incluyen la regresión de las
habilidades motoras, la dificultad de la marcha, ataxia, hipotonía, respuestas
plantares extensores, atrofia óptica, y neuropatía periférica.
• La forma juvenil de MLD presenta entre 3 y 16 años
de edad con trastorno de la marcha, deterioro intelectual, ataxia, signos de la
neurona motora superior, y una neuropatía periférica. Se pueden producir
convulsiones. MLD aparición en el adulto (17 años o más) lo general es
anunciado por la demencia y problemas de conducta. (Ver 'juvenil y del adulto'
más arriba).
● El diagnóstico de LDM se establece mediante la
demostración de la actividad de la
arilsulfatasa (ARSA) deficiente en leucocitos o fibroblastos de piel
cultivados. Sin embargo, el diagnóstico basado únicamente en la actividad de
ARSA se complica por la existencia de pseudodeficiencia de ARSA.
● No existe ningún tratamiento curativo está
actualmente disponible para MLD. Tratamientos de investigación incluyen el
trasplante de médula ósea, terapia génica, o el trasplante de células madre
hematopoyéticas en combinación con la terapia génica.
Fuente de actualización
UpToDate 2017
Referencias
- Wrobe D, Henseler M, Huettler S, et al. Un mutante no glicosilado y funcionalmente deficiente (N215H) de la proteína activador esfingolípido B (SAP-B) en una novela caso de leucodistrofia metacromática (MLD). J Inherit Metab Dis 2000; 23:63.
- Thomas PK, Rey RH, RS Kocen, Brett EM. observaciones ultraestructurales comparativos sobre anormalidades nerviosas periféricas en los finales de las formas de comienzo infantil, juvenil y tardías de leucodistrofia metacromática. Acta Neuropathol 1977; 39: 237.
- Polten A, Fluharty AL, Fluharty CB, et al. Bases moleculares de las diferentes formas de leucodistrofia metacromática. N Engl J Med 1991; 324: 18.
- Berger J, Löschl B, Bernheimer H, et al. Ocurrencia, distribución, y el fenotipo de las mutaciones arilsulfatasa A en pacientes con leucodistrofia metacromática. Am J Med Genet 1997; 69: 335.
- Rauschka H, Cölsch B, Baumann N, et al. leucodistrofia metacromática de inicio tardío: el genotipo influye fuertemente en el fenotipo. Neurología 2006; 67: 859.
- Harvey JS, Carey WF, Morris CP. Importancia de la glicosilación y la poliadenilación variantes en metacromática fenotipo leucodistrofia pseudodeficiency. Hum Mol Genet 1998; 7: 1215.
- Barth ML, Ward, C, Harris A, et al. Frecuencia de arilsulfatasa A pseudodeficiency mutaciones asociadas en una población sana. J Med Genet 1994; 31: 667.
- Gustavson KH, Hagberg B. La incidencia y la genética de leucodistrofia metacromática en el norte de Suecia. Acta Paediatr Scand 1971; 60: 585.
- Von Figura K, Gieselmann V, Jacken J. leucodistrofia metacromática. En: The Metabolic y bases moleculares de la enfermedad hereditaria, octava ed, Scriver CR, Beaudet AL, Sly WS, Valle D. (Eds), McGraw-Hill, Nueva York 2001. p.3695.
- Zlotogora J, Bach G, Barak Y, Elian E. leucodistrofia metacromática en los Judios habbanite: de alta frecuencia en un aislado genético y la detección de heterocigotos. Am J Hum Genet 1980; 32: 663.
- Heinisch U, Zlotogora J, Kafert S, Gieselmann V. Las mutaciones múltiples son responsables de la alta frecuencia de leucodistrofia metacromática en una pequeña área geográfica. Am J Hum Genet 1995; 56:51.
- Holve S, Hu D, McCandless SE. leucodistrofia metacromática en el Navajo: secuelas de las guerras de América y la India del siglo XIX. Am J Med Genet 2001; 101: 203.
- Bonkowsky JL, Nelson C, Kingston JL, et al. La carga de las leucodistrofias hereditarias en los niños. Neurología 2010; 75: 718.
- Malone MJ, Stoffyn P. glicolípidos nervios periféricos en leucodistrofia metacromática. Neurología 1967; 17: 1033.
- Fressinaud C, Vallat JM, Masson M, et al. Del adulto leucodistrofia metacromática se presenta como la neuropatía periférica aislada. Neurología 1992; 42: 1396.
- Cameron CL, Kang PB, Burns TM, et al. ralentización multifocal de la conducción nerviosa en leucodistrofia metacromática. Muscle Nerve 2004; 29: 531.
- Ries M, Deeg KH. Poliposis de la vesícula biliar asociada con leucodistrofia metacromática. Eur J Pediatr 1993; 152: 450.
- Simonovski N, Ackerman Z, Kiderman A, los hallazgos de la vesícula biliar Unusual campos S. en dos hermanos con leucodistrofia metacromática. Radiol Pediatr 1998; 28: 706.
- Cappell MS, Marks M, Kirschenbaum H. hemobilia Massive y colecistitis alitiásica debido a pólipo vesicular benigna. Dig Dis Sci 1993; 38: 1156.
- Garavelli L, Rosato S, Mele A, et al. hemobilia masiva y papilomatosis de la vesícula biliar en la leucodistrofia metacromática: una condición que amenaza la vida. Neuropediatría 2009; 40: 284.
- van Rappard DF, Bugiani M, Boelens JJ, et al. Vesícula biliar y el riesgo de pólipos y carcinoma de leucodistrofia metacromática. Neurología 2016; 87: 103.
- Mahmood A, Berry J, Wenger DA, et al. leucodistrofia metacromática: un caso de trillizos con la variante infantil tardía y una revisión sistemática de la literatura. J Child Neurol 2010; 25: 572.
- Zafeiriou DI, Kontopoulos EE, Michelakakis HM, et al. Neurofisiología y la RM en la leucodistrofia metacromática-infantil tardía. Neurol Pediatr 1999; 21: 843.
- MacFaul R, Cavanagh N, Lago BD, et al. leucodistrofia metacromática: revisión de 38 casos. Arco Dis Child 1982; 57: 168.
- Quigley HA, verde WR. Los estudios histopatológicos clínicos oculares y ultraestructurales de leucodistrofia metacromática del adulto. Am J Ophthalmol 1976; 82: 472.
- Takakura H, Nakano C, Kasagi S, et al. Multimodalidad potenciales evocados en la progresión de la leucodistrofia metacromática. Cerebro Dev 1985; 7: 424.
- Wulff CH, Trojaborg W. adulto leucodistrofia metacromática: hallazgos neurofisiológicos. Neurología 1985; 35: 1776.
- AD Dayan. La neuropatía periférica de leucodistrofia metacromática: observaciones sobre la desmielinización segmentaria y remielinización y la distribución intracelular de sulfátido. J Neurol Neurosurg Psychiatry 1967; 30: 311.
- Luijten JA, Straks W, Blikkendaal-Lieftinck LF, et al. leucodistrofia metacromática: un estudio comparativo de los resultados ultraestructurales en el sistema nervioso periférico de los tres casos, uno de los infantil tardía, uno de los menores y uno de la forma adulta de la enfermedad. Neuropadiatrie 1978; 9: 338.
- Schiffmann R, van der Knaap MS. Artículo invitado: un enfoque basado en la resonancia magnética para el diagnóstico de los trastornos de la materia blanca. Neurología 2009; 72: 750.
- i Dali C, Hanson LG, Barton NW, et al. Brain N-acetil aspartato niveles se correlacionan con la función motora en leucodistrofia metacromática. Neurología 2010; 75: 1896.
- M, Martin Da, Andersson C, et al. TCM hematopoyéticas: un tratamiento útil para fines de leucodistrofia metacromática. Bone Marrow Transplant 2014; 49: 1046.
- de Hosson LD, van de Warrenburg BP, PREIJERS FW, et al. Adulto leucodistrofia metacromática tratado por alo-SCT y una revisión de la literatura. Bone Marrow Transplant 2011; 46: 1071.
- Boucher AA, Miller W, Shanley R, et al. Los resultados a largo plazo después del trasplante alogénico de células madre hematopoyéticas de leucodistrofia metacromática: el mayor informe de cohortes de una sola institución. Orphanet J Rare Dis 2015; 10:94.
- Groeschel S, Kühl JS, Bley AE, et al. Resultado a largo plazo del trasplante alogénico hematopoyéticas Trasplante de células madre en pacientes con menores leucodistrofia metacromática En comparación con los pacientes de control no trasplantados. JAMA Neurol 2016; 73: 1133.
- Batzios SP, Zafeiriou DI. El desarrollo de opciones de tratamiento para leucodistrofia metacromática. Mol Genet Metab 2012; 105: 56.
- Biffi A, Montini E, Lorioli L, et al. Lentivirales hematopoyéticas de genes de células madre beneficios de terapia leucodistrofia metacromática. Ciencia 2013; 341: 1.233.158.
- Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral terapia hematopoyética gen de células madre en leucodistrofia metacromática de inicio temprano: un análisis ad-hoc de un, de etiqueta abierta no aleatorizado, de fase medio de prueba. The Lancet 2016; 388: 476.
No hay comentarios:
Publicar un comentario